Gene expression

Genes are expressed by being transcribed into RNA, and this transcript may then be translated into protein.
Gene expression is the process by which information from a gene is used in the synthesis of a functional gene product. These products are often proteins, but in non-protein coding genes such as transfer RNA (tRNA) or small nuclear RNA (snRNA) genes, the product is a functional RNA.
The process of gene expression is used by all known life—eukaryotes (including multicellular organisms), prokaryotes (bacteria and archaea), and utilized by viruses—to generate the macromolecular machinery for life.
Several steps in the gene expression process may be modulated, including the transcription, RNA splicing, translation, and post-translational modification of a protein. Gene regulation gives the cell control over structure and function, and is the basis for cellular differentiation, morphogenesis and the versatility and adaptability of any organism. Gene regulation may also serve as a substrate for evolutionary change, since control of the timing, location, and amount of gene expression can have a profound effect on the functions (actions) of the gene in a cell or in a multicellular organism.
In genetics, gene expression is the most fundamental level at which the genotype gives rise to the phenotype, i.e. observable trait. The genetic code stored in DNA is "interpreted" by gene expression, and the properties of the expression give rise to the organism's phenotype. Such phenotypes are often expressed by the synthesis of proteins that control the organism's shape, or that act as enzymes catalysing specific metabolic pathways characterising the organism. Regulation of gene expression is thus critical to an organism's development.
Contents
1 Mechanism
1.1 Transcription
1.2 RNA processing
1.3 Non-coding RNA maturation
1.4 RNA export
1.5 Translation
1.6 Folding
1.7 Translocation
1.8 Protein transport
2 Regulation of gene expression
2.1 Transcriptional regulation
2.2 Transcriptional regulation in cancer
2.3 Post-transcriptional regulation
2.4 Three prime untranslated regions and microRNAs
2.5 Translational regulation
2.6 Protein degradation
3 Measurement
3.1 mRNA quantification
3.2 RNA profiles in Wikipedia
3.3 Protein quantification
3.4 Localisation
4 Expression system
4.1 Inducible expression
4.2 In nature
5 Gene networks
6 Techniques and tools
7 Gene expression databases
8 See also
9 References
10 External links
Mechanism
Transcription

The process of transcription is carried out by RNA polymerase (RNAP), Which uses DNA (black) as a template and produces RNA (blue).
A gene is a stretch of DNA that encodes information. Genomic DNA consists of two antiparallel and reverse complementary strands, each having 5' and 3' ends. With respect to a gene, the two strands may be labeled the "template strand," which serves as a blueprint for the production of an RNA transcript, and the "coding strand," which includes the DNA version of the transcript sequence. (Perhaps surprisingly, the "coding strand" is not physically involved in the coding process because it is the "template strand" that is read during transcription.)
The production of the RNA copy of the DNA is called transcription, and is performed in the nucleus by RNA polymerase, which adds one RNA nucleotide at a time to a growing RNA strand as per the complementarity law of the bases. This RNA is complementary to the template 3' → 5' DNA strand,[1] which is itself complementary to the coding 5' → 3' DNA strand. Therefore, the resulting 5' → 3' RNA strand is identical to the coding DNA strand with the exception that Thymines are replaced with uracils (U) in the RNA. A coding DNA strand reading "ATG" is indirectly transcribed through the non-coding strand as "UAC" in RNA.
In prokaryotes, transcription is carried out by a single type of RNA polymerase, which needs a DNA sequence called a Pribnow box as well as a sigma factor (σ factor) to start transcription. In eukaryotes, transcription is performed by three types of RNA polymerases, each of which needs a special DNA sequence called the promoter and a set of DNA-binding proteins—transcription factors—to initiate the process. RNA polymerase I is responsible for transcription of ribosomal RNA (rRNA) genes. RNA polymerase II (Pol II) transcribes all protein-coding genes but also some non-coding RNAs (e.g., snRNAs, snoRNAs or long non-coding RNAs). Pol II includes a C-terminal domain (CTD) that is rich in serine residues. When these residues are phosphorylated, the CTD binds to various protein factors that promote transcript maturation and modification. RNA polymerase III transcribes 5S rRNA, transfer RNA (tRNA) genes, and some small non-coding RNAs (e.g., 7SK). Transcription ends when the polymerase encounters a sequence called the terminator.
RNA processing
While transcription of prokaryotic protein-coding genes creates messenger RNA (mRNA) that is ready for translation into protein, transcription of eukaryotic genes leaves a primary transcript of RNA (pre-mRNA), which first has to undergo a series of modifications to become a mature mRNA.
These include 5' capping, which is set of enzymatic reactions that add 7-methylguanosine (m7G) to the 5' end of pre-mRNA and thus protect the RNA from degradation by exonucleases. The m7G cap is then bound by cap binding complex heterodimer (CBC20/CBC80), which aids in mRNA export to cytoplasm and also protect the RNA from decapping.
Another modification is 3' cleavage and polyadenylation. They occur if polyadenylation signal sequence (5'- AAUAAA-3') is present in pre-mRNA, which is usually between protein-coding sequence and terminator. The pre-mRNA is first cleaved and then a series of ~200 adenines (A) are added to form poly(A) tail, which protects the RNA from degradation. Poly(A) tail is bound by multiple poly(A)-binding proteins (PABP) necessary for mRNA export and translation re-initiation.

Simple illustration of exons and introns in pre-mRNA and the formation of mature mRNA by splicing. The UTRs are non-coding parts of exons at the ends of the mRNA.
A very important modification of eukaryotic pre-mRNA is RNA splicing. The majority of eukaryotic pre-mRNAs consist of alternating segments called exons and introns. During the process of splicing, an RNA-protein catalytical complex known as spliceosome catalyzes two transesterification reactions, which remove an intron and release it in form of lariat structure, and then splice neighbouring exons together. In certain cases, some introns or exons can be either removed or retained in mature mRNA. This so-called alternative splicing creates series of different transcripts originating from a single gene. Because these transcripts can be potentially translated into different proteins, splicing extends the complexity of eukaryotic gene expression.
Extensive RNA processing may be an evolutionary advantage made possible by the nucleus of eukaryotes. In prokaryotes, transcription and translation happen together, whilst in eukaryotes, the nuclear membrane separates the two processes, giving time for RNA processing to occur.
Non-coding RNA maturation
In most organisms non-coding genes (ncRNA) are transcribed as precursors that undergo further processing. In the case of ribosomal RNAs (rRNA), they are often transcribed as a pre-rRNA that contains one or more rRNAs. The pre-rRNA is cleaved and modified (2′-O-methylation and pseudouridine formation) at specific sites by approximately 150 different small nucleolus-restricted RNA species, called snoRNAs. SnoRNAs associate with proteins, forming snoRNPs. While snoRNA part basepair with the target RNA and thus position the modification at a precise site, the protein part performs the catalytical reaction. In eukaryotes, in particular a snoRNP called RNase, MRP cleaves the 45S pre-rRNA into the 28S, 5.8S, and 18S rRNAs. The rRNA and RNA processing factors form large aggregates called the nucleolus.[2]
In the case of transfer RNA (tRNA), for example, the 5' sequence is removed by RNase P,[3] whereas the 3' end is removed by the tRNase Z enzyme[4] and the non-templated 3' CCA tail is added by a nucleotidyl transferase.[5] In the case of micro RNA (miRNA), miRNAs are first transcribed as primary transcripts or pri-miRNA with a cap and poly-A tail and processed to short, 70-nucleotide stem-loop structures known as pre-miRNA in the cell nucleus by the enzymes Drosha and Pasha. After being exported, it is then processed to mature miRNAs in the cytoplasm by interaction with the endonuclease Dicer, which also initiates the formation of the RNA-induced silencing complex (RISC), composed of the Argonaute protein.
Even snRNAs and snoRNAs themselves undergo series of modification before they become part of functional RNP complex. This is done either in the nucleoplasm or in the specialized compartments called Cajal bodies. Their bases are methylated or pseudouridinilated by a group of small Cajal body-specific RNAs (scaRNAs), which are structurally similar to snoRNAs.
RNA export
In eukaryotes most mature RNA must be exported to the cytoplasm from the nucleus. While some RNAs function in the nucleus, many RNAs are transported through the nuclear pores and into the cytosol. Notably this includes all RNA types involved in protein synthesis.[6] In some cases RNAs are additionally transported to a specific part of the cytoplasm, such as a synapse; they are then towed by motor proteins that bind through linker proteins to specific sequences (called "zipcodes") on the RNA.[7]
Translation

During the translation, tRNA charged with amino acid enters the ribosome and aligns with the correct mRNA triplet. Ribosome then adds amino acid to growing protein chain.
For some RNA (non-coding RNA) the mature RNA is the final gene product.[8] In the case of messenger RNA (mRNA) the RNA is an information carrier coding for the synthesis of one or more proteins. mRNA carrying a single protein sequence (common in eukaryotes) is monocistronic whilst mRNA carrying multiple protein sequences (common in prokaryotes) is known as polycistronic.
Every mRNA consists of three parts: a 5' untranslated region (5'UTR), a protein-coding region or open reading frame (ORF), and a 3' untranslated region (3'UTR). The coding region carries information for protein synthesis encoded by the genetic code to form triplets. Each triplet of nucleotides of the coding region is called a codon and corresponds to a binding site complementary to an anticodon triplet in transfer RNA. Transfer RNAs with the same anticodon sequence always carry an identical type of amino acid. Amino acids are then chained together by the ribosome according to the order of triplets in the coding region. The ribosome helps transfer RNA to bind to messenger RNA and takes the amino acid from each transfer RNA and makes a structure-less protein out of it.[9][10] Each mRNA molecule is translated into many protein molecules, on average ~2800 in mammals.[11][12]
In prokaryotes translation generally occurs at the point of transcription (co-transcriptionally), often using a messenger RNA that is still in the process of being created. In eukaryotes translation can occur in a variety of regions of the cell depending on where the protein being written is supposed to be. Major locations are the cytoplasm for soluble cytoplasmic proteins and the membrane of the endoplasmic reticulum for proteins that are for export from the cell or insertion into a cell membrane. Proteins that are supposed to be expressed at the endoplasmic reticulum are recognised part-way through the translation process. This is governed by the signal recognition particle—a protein that binds to the ribosome and directs it to the endoplasmic reticulum when it finds a signal peptide on the growing (nascent) amino acid chain.[13]
Folding
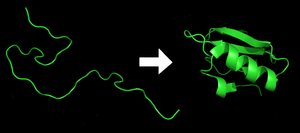
Protein before (left) and after (right) folding
The polypeptide folds into its characteristic and functional three-dimensional structure from a random coil.[14]
Each protein exists as an unfolded polypeptide or random coil when translated from a sequence of mRNA into a linear chain of amino acids. This polypeptide lacks any developed three-dimensional structure (the left hand side of the neighboring figure). Amino acids interact with each other to produce a well-defined three-dimensional structure, the folded protein (the right hand side of the figure) known as the native state. The resulting three-dimensional structure is determined by the amino acid sequence (Anfinsen's dogma).[15]
The correct three-dimensional structure is essential to function, although some parts of functional proteins may remain unfolded.[16] Failure to fold into the intended shape usually produces inactive proteins with different properties including toxic prions. Several neurodegenerative and other diseases are believed to result from the accumulation of misfolded proteins.[17] Many allergies are caused by the folding of the proteins, for the immune system does not produce antibodies for certain protein structures.[18]
Enzymes called chaperones assist the newly formed protein to attain (fold into) the 3-dimensional structure it needs to function.[19] Similarly, RNA chaperones help RNAs attain their functional shapes.[20] Assisting protein folding is one of the main roles of the endoplasmic reticulum in eukaryotes.
Translocation
Secretory proteins of eukaryotes or prokaryotes must be translocated to enter the secretory pathway. Newly synthesized proteins are directed to the eukaryotic Sec61 or prokaryotic SecYEG translocation channel by signal peptides. The efficiency of protein secretion in eukaryotes is very dependent on the signal peptide which has been used.[21]
Protein transport
Many proteins are destined for other parts of the cell than the cytosol and a wide range of signalling sequences or (signal peptides) are used to direct proteins to where they are supposed to be. In prokaryotes this is normally a simple process due to limited compartmentalisation of the cell. However, in eukaryotes there is a great variety of different targeting processes to ensure the protein arrives at the correct organelle.
Not all proteins remain within the cell and many are exported, for example, digestive enzymes, hormones and extracellular matrix proteins. In eukaryotes the export pathway is well developed and the main mechanism for the export of these proteins is translocation to the endoplasmic reticulum, followed by transport via the Golgi apparatus.[22][23]
Regulation of gene expression

The patchy colours of a tortoiseshell cat are the result of different levels of expression of pigmentation genes in different areas of the skin.
Regulation of gene expression refers to the control of the amount and timing of appearance of the functional product of a gene. Control of expression is vital to allow a cell to produce the gene products it needs when it needs them; in turn, this gives cells the flexibility to adapt to a variable environment, external signals, damage to the cell, and other stimuli. More generally, gene regulation gives the cell control over all structure and function, and is the basis for cellular differentiation, morphogenesis and the versatility and adaptability of any organism.
Numerous terms are used to describe types of genes depending on how they are regulated; these include:
- A constitutive gene is a gene that is transcribed continually as opposed to a facultative gene, which is only transcribed when needed.
- A housekeeping gene is a gene that is required to maintain basic cellular function and so is typically expressed in all cell types of an organism. Examples include actin, GAPDH and ubiquitin. Some housekeeping genes are transcribed at a relatively constant rate and these genes can be used as a reference point in experiments to measure the expression rates of other genes.
- A facultative gene is a gene only transcribed when needed as opposed to a constitutive gene.
- An inducible gene is a gene whose expression is either responsive to environmental change or dependent on the position in the cell cycle.
Any step of gene expression may be modulated, from the DNA-RNA transcription step to post-translational modification of a protein. The stability of the final gene product, whether it is RNA or protein, also contributes to the expression level of the gene—an unstable product results in a low expression level. In general gene expression is regulated through changes[24] in the number and type of interactions between molecules[25] that collectively influence transcription of DNA[26] and translation of RNA.[27]
Some simple examples of where gene expression is important are:
- Control of insulin expression so it gives a signal for blood glucose regulation.
X chromosome inactivation in female mammals to prevent an "overdose" of the genes it contains.
Cyclin expression levels control progression through the eukaryotic cell cycle.
Transcriptional regulation

When lactose is present in a prokaryote, it acts as an inducer and inactivates the repressor so that the genes for lactose metabolism can be transcribed.
Regulation of transcription can be broken down into three main routes of influence; genetic (direct interaction of a control factor with the gene), modulation interaction of a control factor with the transcription machinery and epigenetic (non-sequence changes in DNA structure that influence transcription).

The lambda repressor transcription factor (green) binds as a dimer to major groove of DNA target (red and blue) and disables initiation of transcription. From PDB: 1LMB.
Direct interaction with DNA is the simplest and the most direct method by which a protein changes transcription levels. Genes often have several protein binding sites around the coding region with the specific function of regulating transcription. There are many classes of regulatory DNA binding sites known as enhancers, insulators and silencers. The mechanisms for regulating transcription are very varied, from blocking key binding sites on the DNA for RNA polymerase to acting as an activator and promoting transcription by assisting RNA polymerase binding.
The activity of transcription factors is further modulated by intracellular signals causing protein post-translational modification including phosphorylated, acetylated, or glycosylated. These changes influence a transcription factor's ability to bind, directly or indirectly, to promoter DNA, to recruit RNA polymerase, or to favor elongation of a newly synthesized RNA molecule.
The nuclear membrane in eukaryotes allows further regulation of transcription factors by the duration of their presence in the nucleus, which is regulated by reversible changes in their structure and by binding of other proteins.[28] Environmental stimuli or endocrine signals[29] may cause modification of regulatory proteins[30] eliciting cascades of intracellular signals,[31] which result in regulation of gene expression.
More recently it has become apparent that there is a significant influence of non-DNA-sequence specific effects on transcription. These effects are referred to as epigenetic and involve the higher order structure of DNA, non-sequence specific DNA binding proteins and chemical modification of DNA. In general epigenetic effects alter the accessibility of DNA to proteins and so modulate transcription.

In eukaryotes, DNA is organized in form of nucleosomes. Note how the DNA (blue and green) is tightly wrapped around the protein core made of histone octamer (ribbon coils), restricting access to the DNA. From PDB: 1KX5.
DNA methylation is a widespread mechanism for epigenetic influence on gene expression and is seen in bacteria and eukaryotes and has roles in heritable transcription silencing and transcription regulation. In eukaryotes the structure of chromatin, controlled by the histone code, regulates access to DNA with significant impacts on the expression of genes in euchromatin and heterochromatin areas.
Transcriptional regulation in cancer
The majority of gene promoters contain a CpG island with numerous CpG sites.[32] When many of a gene's promoter CpG sites are methylated the gene becomes silenced.[33] Colorectal cancers typically have 3 to 6 driver mutations and 33 to 66 hitchhiker or passenger mutations.[34] However, transcriptional silencing may be of more importance than mutation in causing progression to cancer. For example, in colorectal cancers about 600 to 800 genes are transcriptionally silenced by CpG island methylation (see regulation of transcription in cancer). Transcriptional repression in cancer can also occur by other epigenetic mechanisms, such as altered expression of microRNAs.[35] In breast cancer, transcriptional repression of BRCA1 may occur more frequently by over-expressed microRNA-182 than by hypermethylation of the BRCA1 promoter (see Low expression of BRCA1 in breast and ovarian cancers).
Post-transcriptional regulation
In eukaryotes, where export of RNA is required before translation is possible, nuclear export is thought to provide additional control over gene expression. All transport in and out of the nucleus is via the nuclear pore and transport is controlled by a wide range of importin and exportin proteins.
Expression of a gene coding for a protein is only possible if the messenger RNA carrying the code survives long enough to be translated. In a typical cell, an RNA molecule is only stable if specifically protected from degradation. RNA degradation has particular importance in regulation of expression in eukaryotic cells where mRNA has to travel significant distances before being translated. In eukaryotes, RNA is stabilised by certain post-transcriptional modifications, particularly the 5' cap and poly-adenylated tail.
Intentional degradation of mRNA is used not just as a defence mechanism from foreign RNA (normally from viruses) but also as a route of mRNA destabilisation. If an mRNA molecule has a complementary sequence to a small interfering RNA then it is targeted for destruction via the RNA interference pathway.
Three prime untranslated regions and microRNAs
Three prime untranslated regions (3'UTRs) of messenger RNAs (mRNAs) often contain regulatory sequences that post-transcriptionally influence gene expression. Such 3'-UTRs often contain both binding sites for microRNAs (miRNAs) as well as for regulatory proteins. By binding to specific sites within the 3'-UTR, miRNAs can decrease gene expression of various mRNAs by either inhibiting translation or directly causing degradation of the transcript. The 3'-UTR also may have silencer regions that bind repressor proteins that inhibit the expression of a mRNA.
The 3'-UTR often contains microRNA response elements (MREs). MREs are sequences to which miRNAs bind. These are prevalent motifs within 3'-UTRs. Among all regulatory motifs within the 3'-UTRs (e.g. including silencer regions), MREs make up about half of the motifs.
As of 2014, the miRBase web site,[36] an archive of miRNA sequences and annotations, listed 28,645 entries in 233 biologic species. Of these, 1,881 miRNAs were in annotated human miRNA loci. miRNAs were predicted to have an average of about four hundred target mRNAs (affecting expression of several hundred genes).[37] Freidman et al.[37] estimate that >45,000 miRNA target sites within human mRNA 3'UTRs are conserved above background levels, and >60% of human protein-coding genes have been under selective pressure to maintain pairing to miRNAs.
Direct experiments show that a single miRNA can reduce the stability of hundreds of unique mRNAs.[38] Other experiments show that a single miRNA may repress the production of hundreds of proteins, but that this repression often is relatively mild (less than 2-fold).[39][40]
The effects of miRNA dysregulation of gene expression seem to be important in cancer.[41] For instance, in gastrointestinal cancers, nine miRNAs have been identified as epigenetically altered and effective in down regulating DNA repair enzymes.[42]
The effects of miRNA dysregulation of gene expression also seem to be important in neuropsychiatric disorders, such as schizophrenia, bipolar disorder, major depression, Parkinson's disease, Alzheimer's disease and autism spectrum disorders.[43][44][45]
Translational regulation
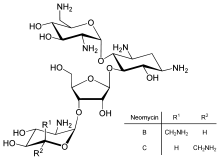
Neomycin is an example of a small molecule that reduces expression of all protein genes inevitably leading to cell death; it thus acts as an antibiotic.
Direct regulation of translation is less prevalent than control of transcription or mRNA stability but is occasionally used. Inhibition of protein translation is a major target for toxins and antibiotics, so they can kill a cell by overriding its normal gene expression control. Protein synthesis inhibitors include the antibiotic neomycin and the toxin ricin.
Protein degradation
Once protein synthesis is complete, the level of expression of that protein can be reduced by protein degradation. There are major protein degradation pathways in all prokaryotes and eukaryotes, of which the proteasome is a common component. An unneeded or damaged protein is often labeled for degradation by addition of ubiquitin.
Measurement
Measuring gene expression is an important part of many life sciences, as the ability to quantify the level at which a particular gene is expressed within a cell, tissue or organism can provide a lot of valuable information. For example, measuring gene expression can:
- Identify viral infection of a cell (viral protein expression).
- Determine an individual's susceptibility to cancer (oncogene expression).
- Find if a bacterium is resistant to penicillin (beta-lactamase expression).
Similarly, the analysis of the location of protein expression is a powerful tool, and this can be done on an organismal or cellular scale. Investigation of localization is particularly important for the study of development in multicellular organisms and as an indicator of protein function in single cells. Ideally, measurement of expression is done by detecting the final gene product (for many genes, this is the protein); however, it is often easier to detect one of the precursors, typically mRNA and to infer gene-expression levels from these measurements.
mRNA quantification
Levels of mRNA can be quantitatively measured by northern blotting, which provides size and sequence information about the mRNA molecules. A sample of RNA is separated on an agarose gel and hybridized to a radioactively labeled RNA probe that is complementary to the target sequence. The radiolabeled RNA is then detected by an autoradiograph. Because the use of radioactive reagents makes the procedure time consuming and potentially dangerous, alternative labeling and detection methods, such as digoxigenin and biotin chemistries, have been developed. Perceived disadvantages of Northern blotting are that large quantities of RNA are required and that quantification may not be completely accurate, as it involves measuring band strength in an image of a gel. On the other hand, the additional mRNA size information from the Northern blot allows the discrimination of alternately spliced transcripts.
Another approach for measuring mRNA abundance is RT-qPCR. In this technique, reverse transcription is followed by quantitative PCR. Reverse transcription first generates a DNA template from the mRNA; this single-stranded template is called cDNA. The cDNA template is then amplified in the quantitative step, during which the fluorescence emitted by labeled hybridization probes or intercalating dyes changes as the DNA amplification process progresses. With a carefully constructed standard curve, qPCR can produce an absolute measurement of the number of copies of original mRNA, typically in units of copies per nanolitre of homogenized tissue or copies per cell. qPCR is very sensitive (detection of a single mRNA molecule is theoretically possible), but can be expensive depending on the type of reporter used; fluorescently labeled oligonucleotide probes are more expensive than non-specific intercalating fluorescent dyes.
For expression profiling, or high-throughput analysis of many genes within a sample, quantitative PCR may be performed for hundreds of genes simultaneously in the case of low-density arrays. A second approach is the hybridization microarray. A single array or "chip" may contain probes to determine transcript levels for every known gene in the genome of one or more organisms. Alternatively, "tag based" technologies like Serial analysis of gene expression (SAGE) and RNA-Seq, which can provide a relative measure of the cellular concentration of different mRNAs, can be used. An advantage of tag-based methods is the "open architecture", allowing for the exact measurement of any transcript, with a known or unknown sequence. Next-generation sequencing (NGS) such as RNA-Seq is another approach, producing vast quantities of sequence data that can be matched to a reference genome. Although NGS is comparatively time-consuming, expensive, and resource-intensive, it can identify single-nucleotide polymorphisms, splice-variants, and novel genes, and can also be used to profile expression in organisms for which little or no sequence information is available.
RNA profiles in Wikipedia
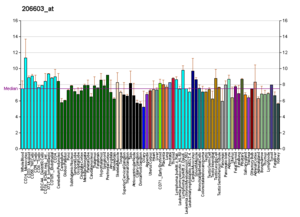
The RNA expression profile of the GLUT4 Transporter (one of the main glucose transporters found in the human body
Profiles like these are found for almost all proteins listed in Wikipedia. They are generated by organizations such as the Genomics Institute of the Novartis Research Foundation and the European Bioinformatics Institute. Additional information can be found by searching their databases (for an example of the GLUT4 transporter pictured here, see citation).[46] These profiles indicate the level of DNA expression (and hence RNA produced) of a certain protein in a certain tissue, and are color-coded accordingly in the images located in the Protein Box on the right side of each Wikipedia page.
Protein quantification
For genes encoding proteins, the expression level can be directly assessed by a number of methods with some clear analogies to the techniques for mRNA quantification.
The most commonly used[citation needed] method is to perform a Western blot against the protein of interest—this gives information on the size of the protein in addition to its identity. A sample (often cellular lysate) is separated on a polyacrylamide gel, transferred to a membrane and then probed with an antibody to the protein of interest. The antibody can either be conjugated to a fluorophore or to horseradish peroxidase for imaging and/or quantification. The gel-based nature of this assay makes quantification less accurate, but it has the advantage of being able to identify later modifications to the protein, for example proteolysis or ubiquitination, from changes in size.
Localisation
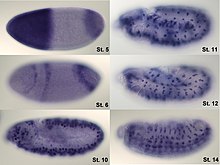
In situ-hybridization of Drosophila embryos at different developmental stages for the mRNA responsible for the expression of hunchback. High intensity of blue color marks places with high hunchback mRNA quantity.
Analysis of expression is not limited to quantification; localisation can also be determined. mRNA can be detected with a suitably labelled complementary mRNA strand and protein can be detected via labelled antibodies. The probed sample is then observed by microscopy to identify where the mRNA or protein is.
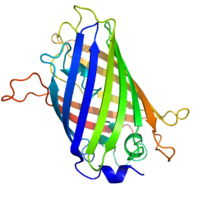
The three-dimensional structure of green fluorescent protein. The residues in the centre of the "barrel" are responsible for production of green light after exposing to higher energetic blue light. From PDB: 1EMA.
By replacing the gene with a new version fused to a green fluorescent protein (or similar) marker, expression may be directly quantified in live cells. This is done by imaging using a fluorescence microscope. It is very difficult to clone a GFP-fused protein into its native location in the genome without affecting expression levels so this method often cannot be used to measure endogenous gene expression. It is, however, widely used to measure the expression of a gene artificially introduced into the cell, for example via an expression vector. It is important to note that by fusing a target protein to a fluorescent reporter the protein's behavior, including its cellular localization and expression level, can be significantly changed.
The enzyme-linked immunosorbent assay works by using antibodies immobilised on a microtiter plate to capture proteins of interest from samples added to the well. Using a detection antibody conjugated to an enzyme or fluorophore the quantity of bound protein can be accurately measured by fluorometric or colourimetric detection. The detection process is very similar to that of a Western blot, but by avoiding the gel steps more accurate quantification can be achieved.
Expression system
Tet-ON inducible shRNA system
An expression system is a system specifically designed for the production of a gene product of choice. This is normally a protein although may also be RNA, such as tRNA or a ribozyme. An expression system consists of a gene, normally encoded by DNA, and the molecular machinery required to transcribe the DNA into mRNA and translate the mRNA into protein using the reagents provided. In the broadest sense this includes every living cell but the term is more normally used to refer to expression as a laboratory tool. An expression system is therefore often artificial in some manner. Expression systems are, however, a fundamentally natural process. Viruses are an excellent example where they replicate by using the host cell as an expression system for the viral proteins and genome.
Inducible expression
Doxycycline is also used in "Tet-on" and "Tet-off" tetracycline controlled transcriptional activation to regulate transgene expression in organisms and cell cultures.
In nature
In addition to these biological tools, certain naturally observed configurations of DNA (genes, promoters, enhancers, repressors) and the associated machinery itself are referred to as an expression system. This term is normally used in the case where a gene or set of genes is switched on under well defined conditions, for example, the simple repressor switch expression system in Lambda phage and the lac operator system in bacteria. Several natural expression systems are directly used or modified and used for artificial expression systems such as the Tet-on and Tet-off expression system.
Gene networks
Genes have sometimes been regarded as nodes in a network, with inputs being proteins such as transcription factors, and outputs being the level of gene expression. The node itself performs a function, and the operation of these functions have been interpreted as performing a kind of information processing within cells and determines cellular behavior.
Gene networks can also be constructed without formulating an explicit causal model. This is often the case when assembling networks from large expression data sets. Covariation and correlation of expression is computed across a large sample of cases and measurements (often transcriptome or proteome data). The source of variation can be either experimental or natural (observational). There are several ways to construct gene expression networks, but one common approach is to compute a matrix of all pair-wise correlations of expression across conditions, time points, or individuals and convert the matrix (after thresholding at some cut-off value) into a graphical representation in which nodes represent genes, transcripts, or proteins and edges connecting these nodes represent the strength of association (see [1]).[47]Weighted correlation network analysis (WGCNA) involves weighted networks defined by soft-thresholding the pairwise correlations among variables (e.g. measures of transcript abundance). WGCNA can be applied to compute eigengenes, which are highly robust biomarkers (features) useful for diagnosis and prognosis.[48]
Techniques and tools
The following experimental techniques are used to measure gene expression and are listed in roughly chronological order, starting with the older, more established technologies. They are divided into two groups based on their degree of multiplexity.
- Low-to-mid-plex techniques:
- Reporter gene
- Northern blot
Western blot[49]
- Fluorescent in situ hybridization
- Reverse transcription PCR
- Higher-plex techniques:
SAGE[50]
DNA microarray[51]
Tiling array[52]
RNA-Seq[53]
Gene expression databases
Gene expression omnibus (GEO) at NCBI[54]
Expression Atlas at the EBI
- Mouse Gene Expression Database at the Jackson Laboratory
CollecTF: a database of experimentally validated transcription factor-binding sites in Bacteria.[55]
- COLOMBOS: collection of bacterial expression compendia.[56]
Many Microbe Microarrays Database: microbial Affymetrix data[57]
See also
 Gene expression portal
Gene expression portal
- AlloMap molecular expression testing
- Bookmarking
- Expressed sequence tag
- Expression Atlas
- Expression profiling
- Gene structure
- Genetic engineering
- Genetically modified organism
- List of biological databases
- List of human genes
- Oscillating gene
- Paramutation
- Protein production
- Protein purification
- Ribonomics
- Ridge
- Sequence profiling tool
- Transcriptional bursting
- Transcriptional noise
References
^ Brueckner F, Armache KJ, Cheung A, Damsma GE, Kettenberger H, Lehmann E, Sydow J, Cramer P (February 2009). "Structure-function studies of the RNA polymerase II elongation complex". Acta Crystallographica D. 65 (Pt 2): 112–20. doi:10.1107/S0907444908039875. PMC 2631633. PMID 19171965..mw-parser-output cite.citation{font-style:inherit}.mw-parser-output q{quotes:"""""""'""'"}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ Sirri V, Urcuqui-Inchima S, Roussel P, Hernandez-Verdun D (January 2008). "Nucleolus: the fascinating nuclear body". Histochemistry and Cell Biology. 129 (1): 13–31. doi:10.1007/s00418-007-0359-6. PMC 2137947. PMID 18046571.
^ Frank DN, Pace NR (1998). "Ribonuclease P: unity and diversity in a tRNA processing ribozyme". Annual Review of Biochemistry. 67: 153–80. doi:10.1146/annurev.biochem.67.1.153. PMID 9759486.
^ Ceballos M, Vioque A (2007). "tRNase Z". Protein and Peptide Letters. 14 (2): 137–45. doi:10.2174/092986607779816050. PMID 17305600.
^ Weiner AM (October 2004). "tRNA maturation: RNA polymerization without a nucleic acid template". Current Biology. 14 (20): R883–5. doi:10.1016/j.cub.2004.09.069. PMID 15498478.
^ Köhler A, Hurt E (October 2007). "Exporting RNA from the nucleus to the cytoplasm". Nature Reviews. Molecular Cell Biology. 8 (10): 761–73. doi:10.1038/nrm2255. PMID 17786152.
^ Jambhekar A, Derisi JL (May 2007). "Cis-acting determinants of asymmetric, cytoplasmic RNA transport". RNA. 13 (5): 625–42. doi:10.1261/rna.262607. PMC 1852811. PMID 17449729.
^ Amaral PP, Dinger ME, Mercer TR, Mattick JS (March 2008). "The eukaryotic genome as an RNA machine". Science. 319 (5871): 1787–9. Bibcode:2008Sci...319.1787A. doi:10.1126/science.1155472. PMID 18369136.
^ Hansen TM, Baranov PV, Ivanov IP, Gesteland RF, Atkins JF (May 2003). "Maintenance of the correct open reading frame by the ribosome". EMBO Reports. 4 (5): 499–504. doi:10.1038/sj.embor.embor825. PMC 1319180. PMID 12717454.
^ Berk V, Cate JH (June 2007). "Insights into protein biosynthesis from structures of bacterial ribosomes". Current Opinion in Structural Biology. 17 (3): 302–9. doi:10.1016/j.sbi.2007.05.009. PMID 17574829.
^ Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (May 2011). "Global quantification of mammalian gene expression control". Nature. 473 (7347): 337–42. Bibcode:2011Natur.473..337S. doi:10.1038/nature10098. PMID 21593866.
^ Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (March 2013). "Corrigendum: Global quantification of mammalian gene expression control". Nature. 495 (7439): 126–7. Bibcode:2013Natur.495..126S. doi:10.1038/nature11848. PMID 23407496.
^ Hegde RS, Kang SW (July 2008). "The concept of translocational regulation". The Journal of Cell Biology. 182 (2): 225–32. doi:10.1083/jcb.200804157. PMC 2483521. PMID 18644895.
^ Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walters P (2002). "The Shape and Structure of Proteins". Molecular Biology of the Cell; Fourth Edition. New York and London: Garland Science. ISBN 0-8153-3218-1.
^ Anfinsen CB (July 1972). "The formation and stabilization of protein structure". The Biochemical Journal. 128 (4): 737–49. doi:10.1042/bj1280737. PMC 1173893. PMID 4565129.
^ Jeremy M. Berg, John L. Tymoczko, Lubert Stryer; Web content by Neil D. Clarke (2002). "3. Protein Structure and Function". Biochemistry. San Francisco: W. H. Freeman. ISBN 0-7167-4684-0.CS1 maint: Multiple names: authors list (link)
^ Selkoe DJ (December 2003). "Folding proteins in fatal ways". Nature. 426 (6968): 900–4. Bibcode:2003Natur.426..900S. doi:10.1038/nature02264. PMID 14685251.
^ Alberts B, Bray D, Hopkin K, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2010). "Protein Structure and Function". Essential Cell Biology (3rd ed.). New York: Garland Science, Taylor and Francis Group, LLC. pp. 120–170.
^ Hebert DN, Molinari M (October 2007). "In and out of the ER: protein folding, quality control, degradation, and related human diseases". Physiological Reviews. 87 (4): 1377–408. doi:10.1152/physrev.00050.2006. PMID 17928587.
^ Russell R (January 2008). "RNA misfolding and the action of chaperones". Frontiers in Bioscience. 13 (13): 1–20. doi:10.2741/2557. PMC 2610265. PMID 17981525.
^ Kober L, Zehe C, Bode J (April 2013). "Optimized signal peptides for the development of high expressing CHO cell lines". Biotechnology and Bioengineering. 110 (4): 1164–73. doi:10.1002/bit.24776. PMID 23124363.
^ Moreau P, Brandizzi F, Hanton S, Chatre L, Melser S, Hawes C, Satiat-Jeunemaitre B (2007). "The plant ER-Golgi interface: a highly structured and dynamic membrane complex". Journal of Experimental Botany. 58 (1): 49–64. doi:10.1093/jxb/erl135. PMID 16990376.
^ Prudovsky I, Tarantini F, Landriscina M, Neivandt D, Soldi R, Kirov A, Small D, Kathir KM, Rajalingam D, Kumar TK (April 2008). "Secretion without Golgi". Journal of Cellular Biochemistry. 103 (5): 1327–43. doi:10.1002/jcb.21513. PMC 2613191. PMID 17786931.
^ Zaidi SK, Young DW, Choi JY, Pratap J, Javed A, Montecino M, Stein JL, Lian JB, van Wijnen AJ, Stein GS (October 2004). "Intranuclear trafficking: organization and assembly of regulatory machinery for combinatorial biological control". The Journal of Biological Chemistry. 279 (42): 43363–6. doi:10.1074/jbc.R400020200. PMID 15277516.
^ Mattick JS, Amaral PP, Dinger ME, Mercer TR, Mehler MF (January 2009). "RNA regulation of epigenetic processes". BioEssays. 31 (1): 51–9. doi:10.1002/bies.080099. PMID 19154003.
^ Martinez NJ, Walhout AJ (April 2009). "The interplay between transcription factors and microRNAs in genome-scale regulatory networks". BioEssays. 31 (4): 435–45. doi:10.1002/bies.200800212. PMC 3118512. PMID 19274664.
^ Tomilin NV (April 2008). "Regulation of mammalian gene expression by retroelements and non-coding tandem repeats". BioEssays. 30 (4): 338–48. doi:10.1002/bies.20741. PMID 18348251.
^ Veitia RA (November 2008). "One thousand and one ways of making functionally similar transcriptional enhancers". BioEssays. 30 (11–12): 1052–7. doi:10.1002/bies.20849. PMID 18937349.
^ Nguyen T, Nioi P, Pickett CB (May 2009). "The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress". The Journal of Biological Chemistry. 284 (20): 13291–5. doi:10.1074/jbc.R900010200. PMC 2679427. PMID 19182219.
^ Paul S (November 2008). "Dysfunction of the ubiquitin-proteasome system in multiple disease conditions: therapeutic approaches". BioEssays. 30 (11–12): 1172–84. doi:10.1002/bies.20852. PMID 18937370.
^ Los M, Maddika S, Erb B, Schulze-Osthoff K (May 2009). "Switching Akt: from survival signaling to deadly response". BioEssays. 31 (5): 492–5. doi:10.1002/bies.200900005. PMC 2954189. PMID 19319914.
^ Saxonov S, Berg P, Brutlag DL (January 2006). "A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters". Proceedings of the National Academy of Sciences of the United States of America. 103 (5): 1412–7. Bibcode:2006PNAS..103.1412S. doi:10.1073/pnas.0510310103. PMC 1345710. PMID 16432200.
^ Bird A (January 2002). "DNA methylation patterns and epigenetic memory". Genes & Development. 16 (1): 6–21. doi:10.1101/gad.947102. PMID 11782440.
^ Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (March 2013). "Cancer genome landscapes". Science. 339 (6127): 1546–58. Bibcode:2013Sci...339.1546V. doi:10.1126/science.1235122. PMC 3749880. PMID 23539594.
^ Tessitore A, Cicciarelli G, Del Vecchio F, Gaggiano A, Verzella D, Fischietti M, Vecchiotti D, Capece D, Zazzeroni F, Alesse E (2014). "MicroRNAs in the DNA Damage/Repair Network and Cancer". International Journal of Genomics. 2014: 820248. doi:10.1155/2014/820248. PMC 3926391. PMID 24616890.
^ miRBase.org
^ ab Friedman RC, Farh KK, Burge CB, Bartel DP (January 2009). "Most mammalian mRNAs are conserved targets of microRNAs". Genome Research. 19 (1): 92–105. doi:10.1101/gr.082701.108. PMC 2612969. PMID 18955434.
^ Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM (February 2005). "Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs". Nature. 433 (7027): 769–73. Bibcode:2005Natur.433..769L. doi:10.1038/nature03315. PMID 15685193.
^ Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N (September 2008). "Widespread changes in protein synthesis induced by microRNAs". Nature. 455 (7209): 58–63. Bibcode:2008Natur.455...58S. doi:10.1038/nature07228. PMID 18668040.
^ Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP (September 2008). "The impact of microRNAs on protein output". Nature. 455 (7209): 64–71. Bibcode:2008Natur.455...64B. doi:10.1038/nature07242. PMC 2745094. PMID 18668037.
^ Palmero EI, de Campos SG, Campos M, de Souza NC, Guerreiro ID, Carvalho AL, Marques MM (July 2011). "Mechanisms and role of microRNA deregulation in cancer onset and progression". Genetics and Molecular Biology. 34 (3): 363–70. doi:10.1590/S1415-47572011000300001. PMC 3168173. PMID 21931505.
^ Bernstein C, Bernstein H (May 2015). "Epigenetic reduction of DNA repair in progression to gastrointestinal cancer". World Journal of Gastrointestinal Oncology. 7 (5): 30–46. doi:10.4251/wjgo.v7.i5.30. PMC 4434036. PMID 25987950.
^ Maffioletti E, Tardito D, Gennarelli M, Bocchio-Chiavetto L (2014). "Micro spies from the brain to the periphery: new clues from studies on microRNAs in neuropsychiatric disorders". Frontiers in Cellular Neuroscience. 8: 75. doi:10.3389/fncel.2014.00075. PMC 3949217. PMID 24653674.
^ Mellios N, Sur M (2012). "The Emerging Role of microRNAs in Schizophrenia and Autism Spectrum Disorders". Frontiers in Psychiatry. 3: 39. doi:10.3389/fpsyt.2012.00039. PMC 3336189. PMID 22539927.
^ Geaghan M, Cairns MJ (August 2015). "MicroRNA and Posttranscriptional Dysregulation in Psychiatry". Biological Psychiatry. 78 (4): 231–9. doi:10.1016/j.biopsych.2014.12.009. PMID 25636176.
^ "GLUT4 RNA Expression Profile".
^ Chesler EJ, Lu L, Wang J, Williams RW, Manly KF (May 2004). "WebQTL: rapid exploratory analysis of gene expression and genetic networks for brain and behavior". Nature Neuroscience. 7 (5): 485–6. doi:10.1038/nn0504-485. PMID 15114364.
^ Foroushani A, Agrahari R, Docking R, Chang L, Duns G, Hudoba M, Karsan A, Zare H (March 2017). "Large-scale gene network analysis reveals the significance of extracellular matrix pathway and homeobox genes in acute myeloid leukemia: an introduction to the Pigengene package and its applications". BMC Medical Genomics. 10 (1): 16. doi:10.1186/s12920-017-0253-6. PMID 28298217.
^ Song Y, Wang W, Qu X, Sun S (February 2009). "Effects of hypoxia inducible factor-1alpha (HIF-1alpha) on the growth & adhesion in tongue squamous cell carcinoma cells". The Indian Journal of Medical Research. 129 (2): 154–63. PMID 19293442.
^ Hanriot L, Keime C, Gay N, Faure C, Dossat C, Wincker P, Scoté-Blachon C, Peyron C, Gandrillon O (September 2008). "A combination of LongSAGE with Solexa sequencing is well suited to explore the depth and the complexity of transcriptome". BMC Genomics. 9: 418. doi:10.1186/1471-2164-9-418. PMC 2562395. PMID 18796152.
^ Wheelan SJ, Martínez Murillo F, Boeke JD (July 2008). "The incredible shrinking world of DNA microarrays". Molecular bioSystems. 4 (7): 726–32. doi:10.1039/b706237k. PMC 2535915. PMID 18563246.
^ Miyakoshi M, Nishida H, Shintani M, Yamane H, Nojiri H (January 2009). "High-resolution mapping of plasmid transcriptomes in different host bacteria". BMC Genomics. 10: 12. doi:10.1186/1471-2164-10-12. PMC 2642839. PMID 19134166.
^ Denoeud F, Aury JM, Da Silva C, Noel B, Rogier O, Delledonne M, Morgante M, Valle G, Wincker P, Scarpelli C, Jaillon O, Artiguenave F (2008). "Annotating genomes with massive-scale RNA sequencing". Genome Biology. 9 (12): R175. doi:10.1186/gb-2008-9-12-r175. PMC 2646279. PMID 19087247.
^ Clough E, Barrett T (2016). "The Gene Expression Omnibus Database". Methods in Molecular Biology. 1418: 93–110. doi:10.1007/978-1-4939-3578-9_5. PMC 4944384. PMID 27008011.
^ Kiliç S, White ER, Sagitova DM, Cornish JP, Erill I (January 2014). "CollecTF: a database of experimentally validated transcription factor-binding sites in Bacteria". Nucleic Acids Research. 42 (Database issue): D156–60. doi:10.1093/nar/gkt1123. PMC 3965012. PMID 24234444.
^ Moretto M, Sonego P, Dierckxsens N, Brilli M, Bianco L, Ledezma-Tejeida D, et al. (January 2016). "COLOMBOS v3.0: leveraging gene expression compendia for cross-species analyses". Nucleic Acids Research. 44 (D1): D620–3. doi:10.1093/nar/gkv1251. PMC 4702885. PMID 26586805.
^ Faith JJ, Driscoll ME, Fusaro VA, Cosgrove EJ, Hayete B, Juhn FS, et al. (January 2008). "Many Microbe Microarrays Database: uniformly normalized Affymetrix compendia with structured experimental metadata". Nucleic Acids Research. 36 (Database issue): D866–70. doi:10.1093/nar/gkm815. PMC 2238822. PMID 17932051.
External links
- Plant Transcription Factor Database and Plant Transcriptional Regulation Data and Analysis Platform
Gene expression | |||||||
|---|---|---|---|---|---|---|---|
| Introduction to genetics |
| ||||||
| Transcription |
| ||||||
| Translation |
| ||||||
| Regulation |
| ||||||
| Influential people |
| ||||||
| |||||||


