Molecular phylogenetics
This article includes a list of references, but its sources remain unclear because it has insufficient inline citations. (November 2013) (Learn how and when to remove this template message) |
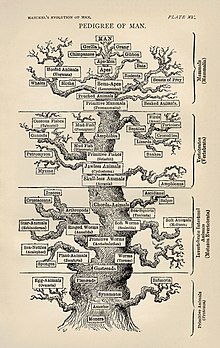
Phylogenetic Tree of Life by Haeckel
Molecular phylogenetics (/məˈlɛkjʊlər ˌfaɪloʊdʒəˈnɛtɪks, mɒ-, moʊ-/[1][2]) is the branch of phylogeny that analyzes genetic, hereditary molecular differences, predominately in DNA sequences, to gain information on an organism's evolutionary relationships. From these analyses, it is possible to determine the processes by which diversity among species has been achieved. The result of a molecular phylogenetic analysis is expressed in a phylogenetic tree. Molecular phylogenetics is one aspect of molecular systematics, a broader term that also includes the use of molecular data in taxonomy and biogeography.
Molecular phylogenetics and molecular evolution correlate. Molecular evolution is the process of selective changes (mutations) at a molecular level (genes, proteins, etc.) throughout various branches in the tree of life (evolution). Molecular phylogenetics makes inferences of the evolutionary relationships that arise due to molecular evolution and results in the construction of a phylogenetic tree. The figure displayed on the right depicts the phylogenetic tree of life as one of the first detailed trees, according to information known in the 1870s by Haeckel.
Contents
1 History
2 Theoretical background
3 Techniques and applications
4 Molecular phylogenetic analysis
5 Limitations
6 See also
7 Notes and references
8 Further reading
9 External links
History
The theoretical frameworks for molecular systematics were laid in the 1960s in the works of Emile Zuckerkandl, Emanuel Margoliash, Linus Pauling, and Walter M. Fitch.[3] Applications of molecular systematics were pioneered by Charles G. Sibley (birds), Herbert C. Dessauer (herpetology), and Morris Goodman (primates), followed by Allan C. Wilson, Robert K. Selander, and John C. Avise (who studied various groups). Work with protein electrophoresis began around 1956. Although the results were not quantitative and did not initially improve on morphological classification, they provided tantalizing hints that long-held notions of the classifications of birds, for example, needed substantial revision. In the period of 1974–1986, DNA-DNA hybridization was the dominant technique used to measure genetic difference.[4]
Theoretical background
Early attempts at molecular systematics were also termed as chemotaxonomy and made use of proteins, enzymes, carbohydrates, and other molecules that were separated and characterized using techniques such as chromatography. These have been replaced in recent times largely by DNA sequencing, which produces the exact sequences of nucleotides or bases in either DNA or RNA segments extracted using different techniques. In general, these are considered superior for evolutionary studies, since the actions of evolution are ultimately reflected in the genetic sequences. At present, it is still a long and expensive process to sequence the entire DNA of an organism (its genome). However, it is quite feasible to determine the sequence of a defined area of a particular chromosome. Typical molecular systematic analyses require the sequencing of around 1000 base pairs. At any location within such a sequence, the bases found in a given position may vary between organisms. The particular sequence found in a given organism is referred to as its haplotype. In principle, since there are four base types, with 1000 base pairs, we could have 41000 distinct haplotypes. However, for organisms within a particular species or in a group of related species, it has been found empirically that only a minority of sites show any variation at all, and most of the variations that are found are correlated, so that the number of distinct haplotypes that are found is relatively small.

In a phylogenetic tree, numerous groupings (clades) exist. A clade may be defined as a group of organisms having a common ancestor throughout evolution. This figure illustrates how a clade in a phylogenetic tree may be expressed.
In a molecular systematic analysis, the haplotypes are determined for a defined area of genetic material; a substantial sample of individuals of the target species or other taxon is used; however, many current studies are based on single individuals. Haplotypes of individuals of closely related, yet different, taxa are also determined. Finally, haplotypes from a smaller number of individuals from a definitely different taxon are determined: these are referred to as an outgroup. The base sequences for the haplotypes are then compared. In the simplest case, the difference between two haplotypes is assessed by counting the number of locations where they have different bases: this is referred to as the number of substitutions (other kinds of differences between haplotypes can also occur, for example the insertion of a section of nucleic acid in one haplotype that is not present in another). The difference between organisms is usually re-expressed as a percentage divergence, by dividing the number of substitutions by the number of base pairs analysed: the hope is that this measure will be independent of the location and length of the section of DNA that is sequenced.
An older and superseded approach was to determine the divergences between the genotypes of individuals by DNA-DNA hybridization. The advantage claimed for using hybridization rather than gene sequencing was that it was based on the entire genotype, rather than on particular sections of DNA. Modern sequence comparison techniques overcome this objection by the use of multiple sequences.
Once the divergences between all pairs of samples have been determined, the resulting triangular matrix of differences is submitted to some form of statistical cluster analysis, and the resulting dendrogram is examined in order to see whether the samples cluster in the way that would be expected from current ideas about the taxonomy of the group. Any group of haplotypes that are all more similar to one another than any of them is to any other haplotype may be said to constitute a clade, which may be visually represented as the figure displayed on the right demonstrates. Statistical techniques such as bootstrapping and jackknifing help in providing reliability estimates for the positions of haplotypes within the evolutionary trees.
Techniques and applications
Every living organism contains deoxyribonucleic acid (DNA), ribonucleic acid (RNA), and proteins. In general, closely related organisms have a high degree of similarity in the molecular structure of these substances, while the molecules of organisms distantly related often show a pattern of dissimilarity. Conserved sequences, such as mitochondrial DNA, are expected to accumulate mutations over time, and assuming a constant rate of mutation, provide a molecular clock for dating divergence. Molecular phylogeny uses such data to build a "relationship tree" that shows the probable evolution of various organisms. With the invention of Sanger sequencing in 1977, it became possible to isolate and identify these molecular structures.[5][6]High-throughput sequencing may also be used to obtain the transcriptome of an organism, allowing inference of phylogenetic relationships using transcriptomic data.
The most common approach is the comparison of homologous sequences for genes using sequence alignment techniques to identify similarity. Another application of molecular phylogeny is in DNA barcoding, wherein the species of an individual organism is identified using small sections of mitochondrial DNA or chloroplast DNA. Another application of the techniques that make this possible can be seen in the very limited field of human genetics, such as the ever-more-popular use of genetic testing to determine a child's paternity, as well as the emergence of a new branch of criminal forensics focused on evidence known as genetic fingerprinting.
Molecular phylogenetic analysis
There are several methods available for performing a molecular phylogenetic analysis. One method, including a comprehensive step-by-step protocol on constructing a phylogenetic tree, including DNA/Amino Acid contiguous sequence assembly, multiple sequence alignment, model-test (testing best-fitting substitution models), and phylogeny reconstruction using Maximum Likelihood and Bayesian Inference, is available at Nature Protocol.[7]
Another molecular phylogenetic analysis technique has been described by Pevsner and shall be summarized in the sentences to follow (Pevsner, 2015). A phylogenetic analysis typically consists of five major steps. The first stage comprises sequence acquisition. The following step consists of performing a multiple sequence alignment, which is the fundamental basis of constructing a phylogenetic tree. The third stage includes different models of DNA and amino acid substitution. Several models of substitution exist. A few examples include Hamming distance, the Jukes and Cantor one-parameter model, and the Kimura two-parameter model (see Models of DNA evolution). The fourth stage consists of various methods of tree building, including distance-based and character-based methods. The normalized Hamming distance and the Jukes-Cantor correction formulas provide the degree of divergence and the probability that a nucleotide changes to another, respectively. Common tree-building methods include unweighted pair group method using arithmetic mean (UPGMA) and Neighbor joining, which are distance-based methods, Maximum parsimony, which is a character-based method, and Maximum likelihood estimation and Bayesian inference, which are character-based/model-based methods. UPGMA is a simple method; however, it is less accurate than the neighbor-joining approach. Finally, the last step comprises evaluating the trees. This assessment of accuracy is composed of consistency, efficiency, and robustness.[8]
Five Stages of Molecular Phylogenetic Analysis
MEGA (molecular evolutionary genetics analysis) is an analysis software that is user-friendly and free to download and use. This software is capable of analyzing both distance-based and character-based tree methodologies. MEGA also contains several options one may choose to utilize, such as heuristic approaches and bootstrapping. Bootstrapping is an approach that is commonly used to measure the robustness of topology in a phylogenetic tree, which demonstrate the percentage each clade is supported after numerous replicates. In general, a value greater than 70% is considered significant. The flow chart displayed on the right visually demonstrates the order of the five stages of Pevsner's molecular phylogenetic analysis technique that have been described.[8]
Limitations
Molecular systematics is an essentially cladistic approach: it assumes that classification must correspond to phylogenetic descent, and that all valid taxa must be monophyletic. This is a limitation when attempting to determine the optimal tree(s), which often involves bisecting and reconnecting portions of the phylogenetic tree(s).
The recent discovery of extensive horizontal gene transfer among organisms provides a significant complication to molecular systematics, indicating that different genes within the same organism can have different phylogenies.
In addition, molecular phylogenies are sensitive to the assumptions and models that go into making them. They face issues such as long-branch attraction, saturation, and taxon sampling problems: This means that strikingly different results can be obtained by applying different models to the same dataset.[9]
Moreover, as previously mentioned, UPGMA is a simple approach in which the tree is always rooted. The algorithm assumes a constant molecular clock for sequences in the tree. This is associated with being a limitation in that if unequal substitution rates exist, the result may be an incorrect tree.[8]
See also
- Computational phylogenetics
- Microbial phylogenetics
- Molecular clock
- Molecular evolution
- PhyloCode
- Phylogenetic nomenclature
Notes and references
^ Jones, Daniel (2003) [1917], Peter Roach, James Hartmann and Jane Setter, eds., English Pronouncing Dictionary, Cambridge: Cambridge University Press, ISBN 3-12-539683-2CS1 maint: Uses editors parameter (link) .mw-parser-output cite.citation{font-style:inherit}.mw-parser-output q{quotes:"""""""'""'"}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ "Phylogenetic". Merriam-Webster Dictionary.
^ Suárez-Díaz, Edna & Anaya-Muñoz, Victor H. (2008). "History, objectivity, and the construction of molecular phylogenies". Stud. Hist. Phil. Biol. & Biomed. Sci. 39 (4): 451–468. doi:10.1016/j.shpsc.2008.09.002. PMID 19026976.
^ Ahlquist, Jon E. (1999). "Charles G. Sibley: A commentary on 30 years of collaboration". The Auk. 116 (3): 856–860. doi:10.2307/4089352. JSTOR 4089352.
^ Sanger F, Coulson AR (May 1975). "A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase". J. Mol. Biol. 94 (3): 441–8. doi:10.1016/0022-2836(75)90213-2. PMID 1100841.
^ Sanger F, Nicklen S, Coulson AR (December 1977). "DNA sequencing with chain-terminating inhibitors". Proc. Natl. Acad. Sci. U.S.A. 74 (12): 5463–7. Bibcode:1977PNAS...74.5463S. doi:10.1073/pnas.74.12.5463. PMC 431765. PMID 271968.
^ Bast, F. (2013). "Sequence Similarity Search, Multiple Sequence Alignment, Model Selection, Distance Matrix and Phylogeny Reconstruction". Protoc. Exch. doi:10.1038/protex.2013.065.
^ abc Pevsner, J. (2015). "Chapter 7: Molecular Phylogeny and Evolution". Bioinformatics and Functional Genomics (3rd ed.). Wiley-Blackwell. pp. 245–295. ISBN 978-1-118-58178-0.
^ Philippe, H.; Brinkmann, H.; Lavrov, D. V.; Littlewood, D. T. J.; Manuel, M.; Wörheide, G.; Baurain, D. (2011). Penny, David, ed. "Resolving Difficult Phylogenetic Questions: Why More Sequences Are Not Enough". PLoS Biology. 9 (3): e1000602. doi:10.1371/journal.pbio.1000602. PMC 3057953. PMID 21423652.
Further reading
Felsenstein, J. 2004. Inferring phylogenies. Sinauer Associates Incorporated.
ISBN 0-87893-177-5.
Hillis, D. M. & Moritz, C. 1996. Molecular systematics. 2nd ed. Sinauer Associates Incorporated.
ISBN 0-87893-282-8.
Page, Roderic D. M.; Holmes, Edward C. (1998). Molecular evolution : a phylogenetic approach. Oxford: Blackwell Science. ISBN 9780865428898. OCLC 47011609.
Page, R. D. M. & Holmes, E. C. 1998. Molecular evolution: a phylogenetic approach. Blackwell Science, Oxford.
ISBN 0-86542-889-1.
Soltis, P.S., Soltis, D.E., and Doyle, J.J. (1992) Molecular systematics of plants. Chapman & Hall, New York.
ISBN 0-41202-231-1.- Soltis, P.S., Soltis, D.E., and Doyle, J.J. (1998) Molecular Systematics of Plants II: DNA Sequencing. Kluwer Academic Publishers Boston, Dordrecht, London.
ISBN 0-41211-131-4.
San Mauro, D.; Agorreta, A. (2010). "Molecular systematics: a synthesis of the common methods and the state of knowledge". Cellular & Molecular Biology Letters. 15 (2): 311–341. doi:10.2478/s11658-010-0010-8.
External links
- NCBI – Systematics and Molecular Phylogenetics
- MEGA Software
- The promise of a DNA taxonomy (Mark L. Blaxter)
Molecular phylogenetics from Encyclopædia Britannica.
Phylogenetics | |
|---|---|
| Relevant fields |
|
| Basic concepts |
|
| Inference methods |
|
| Current topics |
|
| Group traits |
|
| Group types |
|
| Nomenclature |
|
| |
Bioinformatics | |
|---|---|
| Databases |
|
| Software |
|
| Other |
|
| Institutions |
|
| Organizations |
|
| Meetings |
|
| Related topics |
|
| |


