Chemical equilibrium
In a chemical reaction, chemical equilibrium is the state in which both reactants and products are present in concentrations which have no further tendency to change with time, so that there is no observable change in the properties of the system.[1] Usually, this state results when the forward reaction proceeds at the same rate as the reverse reaction. The reaction rates of the forward and backward reactions are generally not zero, but equal. Thus, there are no net changes in the concentrations of the reactant(s) and product(s). Such a state is known as dynamic equilibrium.[2][3]
Contents
1 Historical introduction
2 Thermodynamics
2.1 Addition of reactants or products
2.2 Treatment of activity
2.3 Concentration quotients
2.4 Metastable mixtures
3 Pure substances
4 Multiple equilibria
5 Effect of temperature
6 Effect of electric and magnetic fields
7 Types of equilibrium
8 Composition of a mixture
8.1 Mass-balance equations
8.1.1 Polybasic acids
8.1.2 Solution and precipitation
8.2 Minimization of Gibbs energy
9 See also
10 References
11 Further reading
12 External links
Historical introduction
The concept of chemical equilibrium was developed after Berthollet (1803) found that some chemical reactions are reversible.[4] For any reaction mixture to exist at equilibrium, the rates of the forward and backward (reverse) reactions are equal. In the following chemical equation with arrows pointing both ways to indicate equilibrium,[5] A and B are reactant chemical species, S and T are product species, and α, β, σ, and τ are the stoichiometric coefficients of the respective reactants and products:
α A + β B ⇌ σ S + τ T
The equilibrium concentration position of a reaction is said to lie "far to the right" if, at equilibrium, nearly all the reactants are consumed. Conversely the equilibrium position is said to be "far to the left" if hardly any product is formed from the reactants.
Guldberg and Waage (1865), building on Berthollet’s ideas, proposed the law of mass action:
- forward reaction rate=k+AαBβbackward reaction rate=k−SσTτ{displaystyle {begin{aligned}{text{forward reaction rate}}&=k_{+}{ce {A}}^{alpha }{ce {B}}^{beta }\{text{backward reaction rate}}&=k_{-}{ce {S}}^{sigma }{ce {T}}^{tau }end{aligned}}}

where A, B, S and T are active masses and k+ and k− are rate constants. Since at equilibrium forward and backward rates are equal:
- k+{A}α{B}β=k−{S}σ{T}τ{displaystyle k_{+}left{{ce {A}}right}^{alpha }left{{ce {B}}right}^{beta }=k_{-}left{{ce {S}}right}^{sigma }left{{ce {T}}right}^{tau }}

and the ratio of the rate constants is also a constant, now known as an equilibrium constant.
- Kc=k+k−={S}σ{T}τ{A}α{B}β{displaystyle K_{c}={frac {k_{+}}{k_{-}}}={frac {{{ce {S}}}^{sigma }{{ce {T}}}^{tau }}{{{ce {A}}}^{alpha }{{ce {B}}}^{beta }}}}

By convention the products form the numerator.
However, the law of mass action is valid only for concerted one-step reactions that proceed through a single transition state and is not valid in general because rate equations do not, in general, follow the stoichiometry of the reaction as Guldberg and Waage had proposed (see, for example, nucleophilic aliphatic substitution by SN1 or reaction of hydrogen and bromine to form hydrogen bromide). Equality of forward and backward reaction rates, however, is a necessary condition for chemical equilibrium, though it is not sufficient to explain why equilibrium occurs.
Despite the failure of this derivation, the equilibrium constant for a reaction is indeed a constant, independent of the activities of the various species involved, though it does depend on temperature as observed by the van 't Hoff equation. Adding a catalyst will affect both the forward reaction and the reverse reaction in the same way and will not have an effect on the equilibrium constant. The catalyst will speed up both reactions thereby increasing the speed at which equilibrium is reached.[2][6]
Although the macroscopic equilibrium concentrations are constant in time, reactions do occur at the molecular level. For example, in the case of acetic acid dissolved in water and forming acetate and hydronium ions,
- CH3CO2H + H2O ⇌ CH
3CO−
2 + H3O+
a proton may hop from one molecule of acetic acid on to a water molecule and then on to an acetate anion to form another molecule of acetic acid and leaving the number of acetic acid molecules unchanged. This is an example of dynamic equilibrium. Equilibria, like the rest of thermodynamics, are statistical phenomena, averages of microscopic behavior.
Le Châtelier's principle (1884) predicts the behavior of an equilibrium system when changes to its reaction conditions occur. If a dynamic equilibrium is disturbed by changing the conditions, the position of equilibrium moves to partially reverse the change. For example, adding more S from the outside will cause an excess of products, and the system will try to counteract this by increasing the reverse reaction and pushing the equilibrium point backward (though the equilibrium constant will stay the same).
If mineral acid is added to the acetic acid mixture, increasing the concentration of hydronium ion, the amount of dissociation must decrease as the reaction is driven to the left in accordance with this principle. This can also be deduced from the equilibrium constant expression for the reaction:
- K={CH3CO2−}{H3O+}{CH3CO2H}{displaystyle K={frac {{{ce {CH3CO2-}}}{{ce {H3O+}}}}{{ce {{CH3CO2H}}}}}}

If {H3O+} increases {CH3CO2H} must increase and CH
3CO−
2 must decrease. The H2O is left out, as it is the solvent and its concentration remains high and nearly constant.
A quantitative version is given by the reaction quotient.
J. W. Gibbs suggested in 1873 that equilibrium is attained when the Gibbs free energy of the system is at its minimum value (assuming the reaction is carried out at constant temperature and pressure). What this means is that the derivative of the Gibbs energy with respect to reaction coordinate (a measure of the extent of reaction that has occurred, ranging from zero for all reactants to a maximum for all products) vanishes, signalling a stationary point. This derivative is called the reaction Gibbs energy (or energy change) and corresponds to the difference between the chemical potentials of reactants and products at the composition of the reaction mixture.[1] This criterion is both necessary and sufficient. If a mixture is not at equilibrium, the liberation of the excess Gibbs energy (or Helmholtz energy at constant volume reactions) is the "driving force" for the composition of the mixture to change until equilibrium is reached. The equilibrium constant can be related to the standard Gibbs free energy change for the reaction by the equation
- ΔrG⊖=−RTlnKeq{displaystyle Delta _{r}G^{ominus }=-RTln K_{mathrm {eq} }}

where R is the universal gas constant and T the temperature.
When the reactants are dissolved in a medium of high ionic strength the quotient of activity coefficients may be taken to be constant. In that case the concentration quotient, Kc,
- Kc=[S]σ[T]τ[A]α[B]β{displaystyle K_{ce {c}}={frac {[{ce {S}}]^{sigma }[{ce {T}}]^{tau }}{[{ce {A}}]^{alpha }[{ce {B}}]^{beta }}}}
![{displaystyle K_{ce {c}}={frac {[{ce {S}}]^{sigma }[{ce {T}}]^{tau }}{[{ce {A}}]^{alpha }[{ce {B}}]^{beta }}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c2ac61ea07e9614e7c3e261d737d856396c1b178)
where [A] is the concentration of A, etc., is independent of the analytical concentration of the reactants. For this reason, equilibrium constants for solutions are usually determined in media of high ionic strength. Kc varies with ionic strength, temperature and pressure (or volume). Likewise Kp for gases depends on partial pressure. These constants are easier to measure and encountered in high-school chemistry courses.
Thermodynamics
At constant temperature and pressure, one must consider the Gibbs free energy, G, while at constant temperature and volume, one must consider the Helmholtz free energy: A, for the reaction; and at constant internal energy and volume, one must consider the entropy for the reaction: S.
The constant volume case is important in geochemistry and atmospheric chemistry where pressure variations are significant. Note that, if reactants and products were in standard state (completely pure), then there would be no reversibility and no equilibrium. Indeed, they would necessarily occupy disjoint volumes of space. The mixing of the products and reactants contributes a large entropy (known as entropy of mixing) to states containing equal mixture of products and reactants. The standard Gibbs energy change, together with the Gibbs energy of mixing, determine the equilibrium state.[7][8]
In this article only the constant pressure case is considered. The relation between the Gibbs free energy and the equilibrium constant can be found by considering chemical potentials.[1]
At constant temperature and pressure, the Gibbs free energy, G, for the reaction depends only on the extent of reaction: ξ (Greek letter xi), and can only decrease according to the second law of thermodynamics. It means that the derivative of G with ξ must be negative if the reaction happens; at the equilibrium the derivative being equal to zero.
(dGdξ)T,p=0 {displaystyle left({frac {dG}{dxi }}right)_{T,p}=0~}: equilibrium
In order to meet the thermodynamic condition for equilibrium, the Gibbs energy must be stationary, meaning that the derivative of G with respect to the extent of reaction: ξ, must be zero. It can be shown that in this case, the sum of chemical potentials of the products is equal to the sum of those corresponding to the reactants. Therefore, the sum of the Gibbs energies of the reactants must be the equal to the sum of the Gibbs energies of the products.
- αμA+βμB=σμS+τμT{displaystyle alpha mu _{mathrm {A} }+beta mu _{mathrm {B} }=sigma mu _{mathrm {S} }+tau mu _{mathrm {T} },}

where μ is in this case a partial molar Gibbs energy, a chemical potential. The chemical potential of a reagent A is a function of the activity, {A} of that reagent.
- μA=μA⊖+RTln{A}{displaystyle mu _{mathrm {A} }=mu _{A}^{ominus }+RTln{mathrm {A} },}

(where μo
A is the standard chemical potential).
The definition of the Gibbs energy equation interacts with the fundamental thermodynamic relation to produce
dG=Vdp−SdT+∑i=1kμidNi{displaystyle dG=Vdp-SdT+sum _{i=1}^{k}mu _{i}dN_{i}}.
Inserting dNi = νi dξ into the above equation gives a Stoichiometric coefficient (νi {displaystyle nu _{i}~}
(dGdξ)T,p=∑i=1kμiνi=ΔrGT,p{displaystyle left({frac {dG}{dxi }}right)_{T,p}=sum _{i=1}^{k}mu _{i}nu _{i}=Delta _{mathrm {r} }G_{T,p}}which is the "Gibbs free energy change for the reaction .
This results in:
ΔrGT,p=σμS+τμT−αμA−βμB{displaystyle Delta _{mathrm {r} }G_{T,p}=sigma mu _{mathrm {S} }+tau mu _{mathrm {T} }-alpha mu _{mathrm {A} }-beta mu _{mathrm {B} },}.
By substituting the chemical potentials:
ΔrGT,p=(σμS⊖+τμT⊖)−(αμA⊖+βμB⊖)+(σRTln{S}+τRTln{T})−(αRTln{A}+βRTln{B}){displaystyle Delta _{mathrm {r} }G_{T,p}=(sigma mu _{mathrm {S} }^{ominus }+tau mu _{mathrm {T} }^{ominus })-(alpha mu _{mathrm {A} }^{ominus }+beta mu _{mathrm {B} }^{ominus })+(sigma RTln{mathrm {S} }+tau RTln{mathrm {T} })-(alpha RTln{mathrm {A} }+beta RTln{mathrm {B} })},
the relationship becomes:
- ΔrGT,p=∑i=1kμi⊖νi+RTln{S}σ{T}τ{A}α{B}β{displaystyle Delta _{mathrm {r} }G_{T,p}=sum _{i=1}^{k}mu _{i}^{ominus }nu _{i}+RTln {frac {{mathrm {S} }^{sigma }{mathrm {T} }^{tau }}{{mathrm {A} }^{alpha }{mathrm {B} }^{beta }}}}
∑i=1kμi⊖νi=ΔrG⊖{displaystyle sum _{i=1}^{k}mu _{i}^{ominus }nu _{i}=Delta _{mathrm {r} }G^{ominus }}:

which is the standard Gibbs energy change for the reaction that can be calculated using thermodynamical tables.
The reaction quotient is defined as:
- Qr={S}σ{T}τ{A}α{B}β{displaystyle Q_{mathrm {r} }={frac {{mathrm {S} }^{sigma }{mathrm {T} }^{tau }}{{mathrm {A} }^{alpha }{mathrm {B} }^{beta }}}}

Therefore,
- (dGdξ)T,p=ΔrGT,p=ΔrG⊖+RTlnQr{displaystyle left({frac {dG}{dxi }}right)_{T,p}=Delta _{mathrm {r} }G_{T,p}=Delta _{mathrm {r} }G^{ominus }+RTln Q_{mathrm {r} }}

At equilibrium:
- (dGdξ)T,p=ΔrGT,p=0{displaystyle left({frac {dG}{dxi }}right)_{T,p}=Delta _{mathrm {r} }G_{T,p}=0}

leading to:
- 0=ΔrG⊖+RTlnKeq{displaystyle 0=Delta _{mathrm {r} }G^{ominus }+RTln K_{mathrm {eq} }}

and
- ΔrG⊖=−RTlnKeq{displaystyle Delta _{mathrm {r} }G^{ominus }=-RTln K_{mathrm {eq} }}

Obtaining the value of the standard Gibbs energy change, allows the calculation of the equilibrium constant.
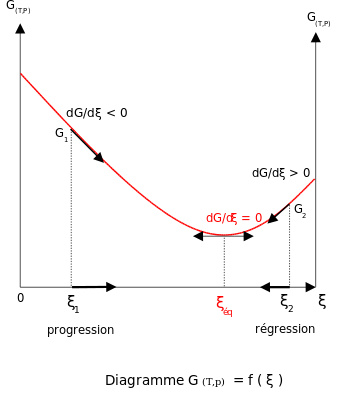
Addition of reactants or products
For a reactional system at equilibrium: Qr = Keq; ξ = ξeq.
- If are modified activities of constituents, the value of the reaction quotient changes and becomes different from the equilibrium constant: Qr ≠ Keq
- (dGdξ)T,p=ΔrG⊖+RTlnQr {displaystyle left({frac {dG}{dxi }}right)_{T,p}=Delta _{mathrm {r} }G^{ominus }+RTln Q_{mathrm {r} }~}
- (dGdξ)T,p=ΔrG⊖+RTlnQr {displaystyle left({frac {dG}{dxi }}right)_{T,p}=Delta _{mathrm {r} }G^{ominus }+RTln Q_{mathrm {r} }~}

- and
- ΔrG⊖=−RTlnKeq {displaystyle Delta _{mathrm {r} }G^{ominus }=-RTln K_{eq}~}
- ΔrG⊖=−RTlnKeq {displaystyle Delta _{mathrm {r} }G^{ominus }=-RTln K_{eq}~}

- then
- (dGdξ)T,p=RTln(QrKeq) {displaystyle left({frac {dG}{dxi }}right)_{T,p}=RTln left({frac {Q_{mathrm {r} }}{K_{mathrm {eq} }}}right)~}
- (dGdξ)T,p=RTln(QrKeq) {displaystyle left({frac {dG}{dxi }}right)_{T,p}=RTln left({frac {Q_{mathrm {r} }}{K_{mathrm {eq} }}}right)~}

- If activity of a reagent i increases
Qr=∏(aj)νj∏(ai)νi {displaystyle Q_{mathrm {r} }={frac {prod (a_{j})^{nu _{j}}}{prod (a_{i})^{nu _{i}}}}~}, the reaction quotient decreases.
- then
Qr<Keq {displaystyle Q_{mathrm {r} }<K_{mathrm {eq} }~}and (dGdξ)T,p<0 {displaystyle left({frac {dG}{dxi }}right)_{T,p}<0~}
- The reaction will shift to the right (i.e. in the forward direction, and thus more products will form).

- If activity of a product j increases
- then
Qr>Keq {displaystyle Q_{mathrm {r} }>K_{mathrm {eq} }~}and (dGdξ)T,p>0 {displaystyle left({frac {dG}{dxi }}right)_{T,p}>0~}
- The reaction will shift to the left (i.e. in the reverse direction, and thus less products will form).

Note that activities and equilibrium constants are dimensionless numbers.
Treatment of activity
The expression for the equilibrium constant can be rewritten as the product of a concentration quotient, Kc and an activity coefficient quotient, Γ.
- K=[S]σ[T]τ...[A]α[B]β...×γSσγTτ...γAαγBβ...=KcΓ{displaystyle K={frac {[mathrm {S} ]^{sigma }[mathrm {T} ]^{tau }...}{[mathrm {A} ]^{alpha }[mathrm {B} ]^{beta }...}}times {frac {{gamma _{mathrm {S} }}^{sigma }{gamma _{mathrm {T} }}^{tau }...}{{gamma _{mathrm {A} }}^{alpha }{gamma _{mathrm {B} }}^{beta }...}}=K_{mathrm {c} }Gamma }
![{displaystyle K={frac {[mathrm {S} ]^{sigma }[mathrm {T} ]^{tau }...}{[mathrm {A} ]^{alpha }[mathrm {B} ]^{beta }...}}times {frac {{gamma _{mathrm {S} }}^{sigma }{gamma _{mathrm {T} }}^{tau }...}{{gamma _{mathrm {A} }}^{alpha }{gamma _{mathrm {B} }}^{beta }...}}=K_{mathrm {c} }Gamma }](https://wikimedia.org/api/rest_v1/media/math/render/svg/160fbc88403a037161d2c9a44d10cd44746215e8)
[A] is the concentration of reagent A, etc. It is possible in principle to obtain values of the activity coefficients, γ. For solutions, equations such as the Debye–Hückel equation or extensions such as Davies equation[9]Specific ion interaction theory or Pitzer equations[10] may be used.Software (below) However this is not always possible. It is common practice to assume that Γ is a constant, and to use the concentration quotient in place of the thermodynamic equilibrium constant. It is also general practice to use the term equilibrium constant instead of the more accurate concentration quotient. This practice will be followed here.
For reactions in the gas phase partial pressure is used in place of concentration and fugacity coefficient in place of activity coefficient. In the real world, for example, when making ammonia in industry, fugacity coefficients must be taken into account. Fugacity, f, is the product of partial pressure and fugacity coefficient. The chemical potential of a species in the gas phase is given by
- μ=μ⊖+RTln(fbar)=μ⊖+RTln(pbar)+RTlnγ{displaystyle mu =mu ^{ominus }+RTln left({frac {f}{mathrm {bar} }}right)=mu ^{ominus }+RTln left({frac {p}{mathrm {bar} }}right)+RTln gamma }

so the general expression defining an equilibrium constant is valid for both solution and gas phases.
Concentration quotients
In aqueous solution, equilibrium constants are usually determined in the presence of an "inert" electrolyte such as sodium nitrate NaNO3 or potassium perchlorate KClO4. The ionic strength of a solution is given by
- I=12∑i=1Ncizi2{displaystyle I={frac {1}{2}}sum _{i=1}^{N}c_{i}z_{i}^{2}}

where ci and zi stand for the concentration and ionic charge of ion type i, and the sum is taken over all the N types of charged species in solution. When the concentration of dissolved salt is much higher than the analytical concentrations of the reagents, the ions originating from the dissolved salt determine the ionic strength, and the ionic strength is effectively constant. Since activity coefficients depend on ionic strength the activity coefficients of the species are effectively independent of concentration. Thus, the assumption that Γ is constant is justified. The concentration quotient is a simple multiple of the equilibrium constant.[11]
- Kc=KΓ{displaystyle K_{mathrm {c} }={frac {K}{Gamma }}}

However, Kc will vary with ionic strength. If it is measured at a series of different ionic strengths the value can be extrapolated to zero ionic strength.[10] The concentration quotient obtained in this manner is known, paradoxically, as a thermodynamic equilibrium constant.
To use a published value of an equilibrium constant in conditions of ionic strength different from the conditions used in its determination, the value should be adjustedSoftware (below).
Metastable mixtures
A mixture may appear to have no tendency to change, though it is not at equilibrium. For example, a mixture of SO2 and O2 is metastable as there is a kinetic barrier to formation of the product, SO3.
- 2 SO2 + O2 ⇌ 2 SO3
The barrier can be overcome when a catalyst is also present in the mixture as in the contact process, but the catalyst does not affect the equilibrium concentrations.
Likewise, the formation of bicarbonate from carbon dioxide and water is very slow under normal conditions
- CO2 + 2 H2O ⇌ HCO−
3 + H3O+
but almost instantaneous in the presence of the catalytic enzyme carbonic anhydrase.
Pure substances
When pure substances (liquids or solids) are involved in equilibria their activities do not appear in the equilibrium constant[12] because their numerical values are considered one.
Applying the general formula for an equilibrium constant to the specific case of a dilute solution of acetic acid in water one obtains
- CH3CO2H + H2O ⇌ CH3CO2− + H3O+
- Kc=[CH3CO2−][H3O+][CH3CO2H][H2O]{displaystyle K_{mathrm {c} }={frac {mathrm {[{CH_{3}CO_{2}}^{-}][{H_{3}O}^{+}]} }{mathrm {[{CH_{3}CO_{2}H}][{H_{2}O}]} }}}
![{displaystyle K_{mathrm {c} }={frac {mathrm {[{CH_{3}CO_{2}}^{-}][{H_{3}O}^{+}]} }{mathrm {[{CH_{3}CO_{2}H}][{H_{2}O}]} }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/53b93bf6dae8354f02ab3e935fca63b9bd4a16c4)
For all but very concentrated solutions, the water can be considered a "pure" liquid, and therefore it has an activity of one. The equilibrium constant expression is therefore usually written as
K=[CH3CO2−][H3O+][CH3CO2H]=Kc{displaystyle K={frac {mathrm {[{CH_{3}CO_{2}}^{-}][{H_{3}O}^{+}]} }{mathrm {[{CH_{3}CO_{2}H}]} }}=K_{mathrm {c} }}.
A particular case is the self-ionization of water itself
- 2 H2O ⇌ H3O+ + OH−
Because water is the solvent, and has an activity of one, the self-ionization constant of water is defined as
- Kw=[H+][OH−]{displaystyle K_{mathrm {w} }=mathrm {[H^{+}][OH^{-}]} }
![{displaystyle K_{mathrm {w} }=mathrm {[H^{+}][OH^{-}]} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/02e72380cadad70b38e1a6f42ce8b217618bc11f)
It is perfectly legitimate to write [H+] for the hydronium ion concentration, since the state of solvation of the proton is constant (in dilute solutions) and so does not affect the equilibrium concentrations. Kw varies with variation in ionic strength and/or temperature.
The concentrations of H+ and OH− are not independent quantities. Most commonly [OH−] is replaced by Kw[H+]−1 in equilibrium constant expressions which would otherwise include hydroxide ion.
Solids also do not appear in the equilibrium constant expression, if they are considered to be pure and thus their activities taken to be one. An example is the Boudouard reaction:[12]
- 2 CO ⇌ CO2 + C
for which the equation (without solid carbon) is written as:
- Kc=[CO2][CO]2{displaystyle K_{mathrm {c} }={frac {mathrm {[CO_{2}]} }{mathrm {[CO]^{2}} }}}
![{displaystyle K_{mathrm {c} }={frac {mathrm {[CO_{2}]} }{mathrm {[CO]^{2}} }}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/df7ff1e7f45707791cfd9dd8e697fe5695e94f60)
Multiple equilibria
Consider the case of a dibasic acid H2A. When dissolved in water, the mixture will contain H2A, HA− and A2−. This equilibrium can be split into two steps in each of which one proton is liberated.
- H2A↽−−⇀HA−+H+:K1=[HA−][H+][H2A]HA−↽−−⇀A2−+H+:K2=[A2−][H+][HA−]{displaystyle {begin{array}{rl}{ce {H2A <=> HA^- + H+}}:&K_{1}={frac {{ce {[HA-][H+]}}}{{ce {[H2A]}}}}\{ce {HA- <=> A^2- + H+}}:&K_{2}={frac {{ce {[A^{2-}][H+]}}}{{ce {[HA-]}}}}end{array}}}
![{displaystyle {begin{array}{rl}{ce {H2A <=> HA^- + H+}}:&K_{1}={frac {{ce {[HA-][H+]}}}{{ce {[H2A]}}}}\{ce {HA- <=> A^2- + H+}}:&K_{2}={frac {{ce {[A^{2-}][H+]}}}{{ce {[HA-]}}}}end{array}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/964f444d4c5ff73dc96c30e9eca7e742a64c7cbc)
K1 and K2 are examples of stepwise equilibrium constants. The overall equilibrium constant, βD, is product of the stepwise constants.
H2A↽−−⇀A2−+2H+{displaystyle {ce {{H2A}<=>{A^{2-}}+{2H+}}}}: βD=[A2−][H+]2[H2A]=K1K2{displaystyle beta _{{ce {D}}}={frac {{ce {[A^{2-}][H^+]^2}}}{{ce {[H_2A]}}}}=K_{1}K_{2}}
![{displaystyle beta _{{ce {D}}}={frac {{ce {[A^{2-}][H^+]^2}}}{{ce {[H_2A]}}}}=K_{1}K_{2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c204101410770f2cc038f6d29c1a7a0da1d363a1)
Note that these constants are dissociation constants because the products on the right hand side of the equilibrium expression are dissociation products. In many systems, it is preferable to use association constants.
- A2−+H+↽−−⇀HA−:β1=[HA−][A2−][H+]A2−+2H+↽−−⇀H2A:β2=[H2A][A2−][H+]2{displaystyle {begin{array}{ll}{ce {A^2- + H+ <=> HA-}}:&beta _{1}={frac {{ce {[HA^-]}}}{{ce {[A^{2-}][H+]}}}}\{ce {A^2- + 2H+ <=> H2A}}:&beta _{2}={frac {{ce {[H2A]}}}{{ce {[A^{2-}][H+]^2}}}}end{array}}}
![{displaystyle {begin{array}{ll}{ce {A^2- + H+ <=> HA-}}:&beta _{1}={frac {{ce {[HA^-]}}}{{ce {[A^{2-}][H+]}}}}\{ce {A^2- + 2H+ <=> H2A}}:&beta _{2}={frac {{ce {[H2A]}}}{{ce {[A^{2-}][H+]^2}}}}end{array}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/266ddb8f43fb05b0d3a3195274e597a4fd3d6f37)
β1 and β2 are examples of association constants. Clearly β1 = 1/K2 and β2 = 1/βD; log β1 = pK2 and log β2 = pK2 + pK1[13]
For multiple equilibrium systems, also see: theory of Response reactions.
Effect of temperature
The effect of changing temperature on an equilibrium constant is given by the van 't Hoff equation
- dlnKdT=ΔHm⊖RT2{displaystyle {frac {dln K}{dT}}={frac {Delta H_{mathrm {m} }^{ominus }}{RT^{2}}}}

Thus, for exothermic reactions (ΔH is negative), K decreases with an increase in temperature, but, for endothermic reactions, (ΔH is positive) K increases with an increase temperature. An alternative formulation is
- dlnKd(T−1)=−ΔHm⊖R{displaystyle {frac {dln K}{d(T^{-1})}}=-{frac {Delta H_{mathrm {m} }^{ominus }}{R}}}

At first sight this appears to offer a means of obtaining the standard molar enthalpy of the reaction by studying the variation of K with temperature. In practice, however, the method is unreliable because error propagation almost always gives very large errors on the values calculated in this way.
Effect of electric and magnetic fields
The effect of electric field on equilibrium has been studied by Manfred Eigen[citation needed] among others.
Types of equilibrium
.mw-parser-output .quotebox{background-color:#F9F9F9;border:1px solid #aaa;box-sizing:border-box;padding:10px;font-size:88%}.mw-parser-output .quotebox.floatleft{margin:0.5em 1.4em 0.8em 0}.mw-parser-output .quotebox.floatright{margin:0.5em 0 0.8em 1.4em}.mw-parser-output .quotebox.centered{margin:0.5em auto 0.8em auto}.mw-parser-output .quotebox.floatleft p,.mw-parser-output .quotebox.floatright p{font-style:inherit}.mw-parser-output .quotebox-title{background-color:#F9F9F9;text-align:center;font-size:larger;font-weight:bold}.mw-parser-output .quotebox-quote.quoted:before{font-family:"Times New Roman",serif;font-weight:bold;font-size:large;color:gray;content:" “ ";vertical-align:-45%;line-height:0}.mw-parser-output .quotebox-quote.quoted:after{font-family:"Times New Roman",serif;font-weight:bold;font-size:large;color:gray;content:" ” ";line-height:0}.mw-parser-output .quotebox .left-aligned{text-align:left}.mw-parser-output .quotebox .right-aligned{text-align:right}.mw-parser-output .quotebox .center-aligned{text-align:center}.mw-parser-output .quotebox cite{display:block;font-style:normal}@media screen and (max-width:360px){.mw-parser-output .quotebox{min-width:100%;margin:0 0 0.8em!important;float:none!important}}
- N2 (g) ⇌ N2 (adsorbed)
- N2 (adsorbed) ⇌ 2 N (adsorbed)
- H2 (g) ⇌ H2 (adsorbed)
- H2 (adsorbed) ⇌ 2 H (adsorbed)
- N (adsorbed) + 3 H(adsorbed) ⇌ NH3 (adsorbed)
- NH3 (adsorbed) ⇌ NH3 (g)
- In the gas phase: rocket engines[14]
- The industrial synthesis such as ammonia in the Haber–Bosch process (depicted right) takes place through a succession of equilibrium steps including adsorption processes
- Atmospheric chemistry
- Seawater and other natural waters: chemical oceanography
- Distribution between two phases
log D distribution coefficient: important for pharmaceuticals where lipophilicity is a significant property of a drug
Liquid–liquid extraction, Ion exchange, Chromatography
- Solubility product
- Uptake and release of oxygen by hemoglobin in blood
- Acid–base equilibria: acid dissociation constant, hydrolysis, buffer solutions, indicators, acid–base homeostasis
- Metal–ligand complexation: sequestering agents, chelation therapy, MRI contrast reagents, Schlenk equilibrium
- Adduct formation: host–guest chemistry, supramolecular chemistry, molecular recognition, dinitrogen tetroxide
- In certain oscillating reactions, the approach to equilibrium is not asymptotically but in the form of a damped oscillation .[12]
- The related Nernst equation in electrochemistry gives the difference in electrode potential as a function of redox concentrations.
- When molecules on each side of the equilibrium are able to further react irreversibly in secondary reactions, the final product ratio is determined according to the Curtin–Hammett principle.
In these applications, terms such as stability constant, formation constant, binding constant, affinity constant, association/dissociation constant are used. In biochemistry, it is common to give units for binding constants, which serve to define the concentration units used when the constant’s value was determined.
Composition of a mixture
When the only equilibrium is that of the formation of a 1:1 adduct as the composition of a mixture, there are many ways that the composition of a mixture can be calculated. For example, see ICE table for a traditional method of calculating the pH of a solution of a weak acid.
There are three approaches to the general calculation of the composition of a mixture at equilibrium.
- The most basic approach is to manipulate the various equilibrium constants until the desired concentrations are expressed in terms of measured equilibrium constants (equivalent to measuring chemical potentials) and initial conditions.
- Minimize the Gibbs energy of the system.[15][16]
- Satisfy the equation of mass balance. The equations of mass balance are simply statements that demonstrate that the total concentration of each reactant must be constant by the law of conservation of mass.
Mass-balance equations
In general, the calculations are rather complicated or complex. For instance, in the case of a dibasic acid, H2A dissolved in water the two reactants can be specified as the conjugate base, A2−, and the proton, H+. The following equations of mass-balance could apply equally well to a base such as 1,2-diaminoethane, in which case the base itself is designated as the reactant A:
- TA=[A]+[HA]+[H2A]{displaystyle T_{mathrm {A} }=mathrm {[A]+[HA]+[H_{2}A]} ,}
- TH=[H]+[HA]+2[H2A]−[OH]{displaystyle T_{mathrm {H} }=mathrm {[H]+[HA]+2[H_{2}A]-[OH]} ,}
![{displaystyle T_{mathrm {A} }=mathrm {[A]+[HA]+[H_{2}A]} ,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b4caf4ff0552bd0430f4b305b3b92c05aef15aa2)
![{displaystyle T_{mathrm {H} }=mathrm {[H]+[HA]+2[H_{2}A]-[OH]} ,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/094ecf5d3e87ba5a02d744835486f24086804cfc)
With TA the total concentration of species A. Note that it is customary to omit the ionic charges when writing and using these equations.
When the equilibrium constants are known and the total concentrations are specified there are two equations in two unknown "free concentrations" [A] and [H]. This follows from the fact that [HA] = β1[A][H], [H2A] = β2[A][H]2 and [OH] = Kw[H]−1
- TA=[A]+β1[A][H]+β2[A][H]2{displaystyle T_{mathrm {A} }=mathrm {[A]} +beta _{1}mathrm {[A][H]} +beta _{2}mathrm {[A][H]} ^{2},}
- TH=[H]+β1[A][H]+2β2[A][H]2−Kw[H]−1{displaystyle T_{mathrm {H} }=mathrm {[H]} +beta _{1}mathrm {[A][H]} +2beta _{2}mathrm {[A][H]} ^{2}-K_{w}[mathrm {H} ]^{-1},}
![{displaystyle T_{mathrm {A} }=mathrm {[A]} +beta _{1}mathrm {[A][H]} +beta _{2}mathrm {[A][H]} ^{2},}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f6ae376d767bfb698f8d771a8aa374366c647255)
![{displaystyle T_{mathrm {H} }=mathrm {[H]} +beta _{1}mathrm {[A][H]} +2beta _{2}mathrm {[A][H]} ^{2}-K_{w}[mathrm {H} ]^{-1},}](https://wikimedia.org/api/rest_v1/media/math/render/svg/25502da2d24e51d7cb6c53782a5392673c4b0819)
so the concentrations of the "complexes" are calculated from the free concentrations and the equilibrium constants.
General expressions applicable to all systems with two reagents, A and B would be
- TA=[A]+∑ipiβi[A]pi[B]qi{displaystyle T_{mathrm {A} }=[mathrm {A} ]+sum _{i}p_{i}beta _{i}[mathrm {A} ]^{p_{i}}[mathrm {B} ]^{q_{i}}}
- TB=[B]+∑iqiβi[A]pi[B]qi{displaystyle T_{mathrm {B} }=[mathrm {B} ]+sum _{i}q_{i}beta _{i}[mathrm {A} ]^{p_{i}}[mathrm {B} ]^{q_{i}}}
![{displaystyle T_{mathrm {A} }=[mathrm {A} ]+sum _{i}p_{i}beta _{i}[mathrm {A} ]^{p_{i}}[mathrm {B} ]^{q_{i}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/de4c20306740d1b3f30387f17ac264b500d313b8)
![{displaystyle T_{mathrm {B} }=[mathrm {B} ]+sum _{i}q_{i}beta _{i}[mathrm {A} ]^{p_{i}}[mathrm {B} ]^{q_{i}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4c9dea321370bf3d5b01364cfed9dbe3c941ce31)
It is easy to see how this can be extended to three or more reagents.
Polybasic acids

Species concentrations during hydrolysis of the aluminium.
The composition of solutions containing reactants A and H is easy to calculate as a function of p[H]. When [H] is known, the free concentration [A] is calculated from the mass-balance equation in A.
The diagram alongside, shows an example of the hydrolysis of the aluminium Lewis acid Al3+(aq)[17] shows the species concentrations for a 5 × 10−6 M solution of an aluminium salt as a function of pH. Each concentration is shown as a percentage of the total aluminium.
Solution and precipitation
The diagram above illustrates the point that a precipitate that is not one of the main species in the solution equilibrium may be formed. At pH just below 5.5 the main species present in a 5 μM solution of Al3+ are aluminium hydroxides Al(OH)2+, AlOH+
2 and Al
13(OH)7+
32, but on raising the pH Al(OH)3 precipitates from the solution. This occurs because Al(OH)3 has a very large lattice energy. As the pH rises more and more Al(OH)3 comes out of solution. This is an example of Le Châtelier's principle in action: Increasing the concentration of the hydroxide ion causes more aluminium hydroxide to precipitate, which removes hydroxide from the solution. When the hydroxide concentration becomes sufficiently high the soluble aluminate, Al(OH)−
4, is formed.
Another common instance where precipitation occurs is when a metal cation interacts with an anionic ligand to form an electrically neutral complex. If the complex is hydrophobic, it will precipitate out of water. This occurs with the nickel ion Ni2+ and dimethylglyoxime, (dmgH2): in this case the lattice energy of the solid is not particularly large, but it greatly exceeds the energy of solvation of the molecule Ni(dmgH)2.
Minimization of Gibbs energy
At equilibrium, at a specified temperature and pressure, the Gibbs energy G is at a minimum:
- dG=∑j=1mμjdNj=0{displaystyle dG=sum _{j=1}^{m}mu _{j},dN_{j}=0}

For a closed system, no particles may enter or leave, although they may combine in various ways. The total number of atoms of each element will remain constant. This means that the minimization above must be subjected to the constraints:
- ∑j=1maijNj=bi0{displaystyle sum _{j=1}^{m}a_{ij}N_{j}=b_{i}^{0}}

where aij is the number of atoms of element i in molecule j and b0
i is the total number of atoms of element i, which is a constant, since the system is closed. If there are a total of k types of atoms in the system, then there will be k such equations. If ions are involved, an additional row is added to the aij matrix specifying the respective charge on each molecule which will sum to zero.
This is a standard problem in optimisation, known as constrained minimisation. The most common method of solving it is using the method of Lagrange multipliers, also known as undetermined multipliers (though other methods may be used).
Define:
- G=G+∑i=1kλi(∑j=1maijNj−bi0)=0{displaystyle {mathcal {G}}=G+sum _{i=1}^{k}lambda _{i}left(sum _{j=1}^{m}a_{ij}N_{j}-b_{i}^{0}right)=0}

where the λi are the Lagrange multipliers, one for each element. This allows each of the Nj and λj to be treated independently, and it can be shown using the tools of multivariate calculus that the equilibrium condition is given by
- 0=∂G∂Nj=μj+∑i=1kλiaij{displaystyle 0={frac {partial {mathcal {G}}}{partial N_{j}}}=mu _{j}+sum _{i=1}^{k}lambda _{i}a_{ij}}
- 0=∂G∂λi=∑j=1maijNj−bi0{displaystyle 0={frac {partial {mathcal {G}}}{partial lambda _{i}}}=sum _{j=1}^{m}a_{ij}N_{j}-b_{i}^{0}}


(For proof see Lagrange multipliers.) This is a set of (m + k) equations in (m + k) unknowns (the Nj and the λi) and may, therefore, be solved for the equilibrium concentrations Nj as long as the chemical potentials are known as functions of the concentrations at the given temperature and pressure. (See Thermodynamic databases for pure substances.) Note that the second equation is just the initial constraints for minimization.
This method of calculating equilibrium chemical concentrations is useful for systems with a large number of different molecules. The use of k atomic element conservation equations for the mass constraint is straightforward, and replaces the use of the stoichiometric coefficient equations.[14] The results are consistent with those specified by chemical equations. For example, if equilibrium is specified by a single chemical equation: [18],
- ∑j=0mνjRj=0{displaystyle sum _{j=0}^{m}nu _{j}R_{j}=0}

where νj is the stochiometric coefficient for the j th molecule (negative for reactants, positive for products) and Rj is the symbol for the j th molecule, a properly balanced equation will obey:
- ∑j=1maijνj=0{displaystyle sum _{j=1}^{m}a_{ij}nu _{j}=0}

Multiplying the first equilibrium condition by νj yields
- 0=∑j=1mνjμj+∑j=1m∑i=1kνjλiaij=∑j=1mνjμj{displaystyle 0=sum _{j=1}^{m}nu _{j}mu _{j}+sum _{j=1}^{m}sum _{i=1}^{k}nu _{j}lambda _{i}a_{ij}=sum _{j=1}^{m}nu _{j}mu _{j}}

As above, defining ΔG
- ΔG=∑j=1mνjμj=∑j=1mνj(μj⊖+RTln({Rj}))=ΔG⊖+RTln(∏j=1m{Rj}νj)=ΔG⊖+RTln(Qr){displaystyle Delta G=sum _{j=1}^{m}nu _{j}mu _{j}=sum _{j=1}^{m}nu _{j}(mu _{j}^{ominus }+RTln({R_{j}}))=Delta G^{ominus }+RTln left(prod _{j=1}^{m}{R_{j}}^{nu _{j}}right)=Delta G^{ominus }+RTln(Q_{r})}

which will be zero at equilibrium.
See also
- Acidosis
- Alkalosis
- Arterial blood gas
- Benesi–Hildebrand method
- Determination of equilibrium constants
- Equilibrium constant
- Henderson–Hasselbalch equation
- Michaelis–Menten kinetics
- pCO2
- pH
- pKa
- Redox equilibria
- Steady state (chemistry)
- Thermodynamic databases for pure substances
Non-random two-liquid model (NRTL model) - Phase equilibrium calculations
UNIQUAC model - Phase equilibrium calculations
References
^ abc Atkins, Peter; De Paula, Julio (2006). Atkins' Physical Chemistry (8th ed.). W. H. Freeman. pp. 200–202. ISBN 0-7167-8759-8..mw-parser-output cite.citation{font-style:inherit}.mw-parser-output q{quotes:"""""""'""'"}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ ab Atkins, Peter W.; Jones, Loretta. Chemical Principles: The Quest for Insight (2nd ed.). ISBN 0-7167-9903-0.
^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "chemical equilibrium".
^ Berthollet, C.L. (1803). Essai de statique chimique [Essay on chemical statics] (in French). Paris, France: Firmin Didot. On pp. 404–407, Berthellot mentions that when he accompanied Napoleon on his expedition to Egypt, he (Berthellot) visited Lake Natron and found sodium carbonate along its shores. He realized that this was a product of the reverse of the usual reaction Na2CO3 + CaCl2 → 2NaCl + CaCO3↓ and therefore that the final state of a reaction was a state of equilibrium between two opposing processes. From p. 405: " … la décomposition du muriate de soude continue donc jusqu'à ce qu'il se soit formé assez de muriate de chaux, parce que l'acide muriatique devant se partager entre les deux bases en raison de leur action, il arrive un terme où leurs forces se balancent." ( … the decomposition of the sodium chloride thus continues until enough calcium chloride is formed, because the hydrochloric acid must be shared between the two bases in the ratio of their action [i.e., capacity to react]; it reaches an end [point] at which their forces are balanced.)
^ The notation ⇌ was proposed in 1884 by the Dutch chemist Jacobus Henricus van 't Hoff. See: van 't Hoff, J.H. (1884). Études de Dynamique Chemique [Studies of chemical dynamics] (in French). Amsterdam, Netherlands: Frederik Muller & Co. pp. 4–5. Van 't Hoff called reactions that didn't proceed to completion "limited reactions". From pp. 4–5: "Or M. Pfaundler a relié ces deux phénomênes … s'accomplit en même temps dans deux sens opposés." (Now Mr. Pfaundler has joined these two phenomena in a single concept by considering the observed limit as the result of two opposing reactions, driving the one in the example cited to the formation of sea salt [i.e., NaCl] and nitric acid, [and] the other to hydrochloric acid and sodium nitrate. This consideration, which experiment validates, justifies the expression "chemical equilibrium", which is used to characterize the final state of limited reactions. I would propose to translate this expression by the following symbol:
- HCl + NO3 Na ⇌ NO3 H + Cl Na .
I thus replace, in this case, the = sign in the chemical equation by the sign ⇌, which in reality doesn't express just equality but shows also the direction of the reaction. This clearly expresses that a chemical action occurs simultaneously in two opposing directions.)
^ Brady, James E. Chemistry: Matter and Its Changes (4th ed.). Fred Senese. ISBN 0-471-21517-1.
^ Schultz, Mary Jane (1999). "Why Equilibrium? Understanding Entropy of Mixing". Journal of Chemical Education. 76 (10): 1391. Bibcode:1999JChEd..76.1391S. doi:10.1021/ed076p1391.
^ Clugston, Michael J. (1990). "A mathematical verification of the second law of thermodynamics from the entropy of mixing". Journal of Chemical Education. 67 (3): 203. Bibcode:1990JChEd..67Q.203C. doi:10.1021/ed067p203.
^ Davies, C. W. (1962). Ion Association. Butterworths.
^ ab Grenthe, I.; Wanner, H. "Guidelines for the extrapolation to zero ionic strength" (PDF).
^ Rossotti, F. J. C.; Rossotti, H. (1961). The Determination of Stability Constants. McGraw-Hill.
^ abc Concise Encyclopedia Chemistry. 1994. ISBN 0-89925-457-8.
^ Beck, M. T.; Nagypál, I. (1990). Chemistry of Complex Equilibria (2nd ed.). Budapest: Akadémiai Kaidó.
^ ab "Computer Program for Calculation of Complex Chemical Equilibrium Compositions and Applications" (PDF). NASA Reference publication 1311. NASA. 1994.
^ Smith, W. R.; Missen, R. W. (1991). Chemical Reaction Equilibrium Analysis: Theory and Algorithms (Reprinted ed.). Malabar, FL: Krieger Publishing.
^ "Mathtrek Systems".
^ The diagram was created with the program HySS
^ C. Kittel, H. Kroemer (1980). "9". Thermal Physics (2 ed.). W. H. Freeman Company. ISBN 0-7167-1088-9.
Further reading
Library resources about Chemical equilibrium |
|
Van Zeggeren, F.; Storey, S. H. (1970). The Computation of Chemical Equilibria. Cambridge University Press. Mainly concerned with gas-phase equilibria.
Leggett, D. J., ed. (1985). Computational Methods for the Determination of Formation Constants. Plenum Press.
Martell, A. E.; Motekaitis, R. J. (1992). The Determination and Use of Stability Constants. Wiley-VCH.
External links
 Media related to Chemical equilibria at Wikimedia Commons
Media related to Chemical equilibria at Wikimedia Commons


